1. Non-standard marketing authorisations
1. Non-standard marketing authorisations
1.1. Conditional Marketing Authorisation (CMA)
For certain categories of medicines, marketing authorisations may be granted on the basis of less complete data than what is normally required, in order to meet unmet medical needs* (as defined within the Article 4 (2) Commission Regulations EC n 507/2006) of patients and in the interest of public health*. In such cases, it is possible for the EMA Committee for Medicinal Products for Human Use (CHMP) to recommend the granting of a marketing authorisation subject to certain specific obligations to be reviewed annually ('conditional marketing authorisation').
*Quote from guideline: “there is no single definition of what constitutes major public health interest. This should be justified by the applicant and assessed by the CHMP on a case-by-case basis. Typically, the justification could present the arguments to support the claim that the medicine addresses to a significant extent the unmet medical needs for maintaining and improving the health of the Community, for example, by introducing new methods of therapy or improves existing ones. It is noted that a new mechanism of action or a technical innovation per se may not necessarily represent a valid argument for justifying major interest from the point of view of public health.”
Medicines that are eligible for centralised approval by the Commission and which:
- aim at the treatment, the prevention or the medical diagnosis of seriously debilitating diseases or life-threatening diseases; or
- are also intended to be used in emergency situations, in response to public threats duly recognised either by the WHO or by the Community for a public health emergency (e.g., a pandemic; see also 1.1.1. Rolling review); or
- are designated as orphan medicinal products in accordance with the Orphan Regulation.
Criteria
A conditional MA may be granted where although comprehensive clinical data referring to the safety and efficacy of the medicine have not been supplied, all the following requirements are met:
- The currently known benefit-risk balance of the medicine is positive.
- It is likely that the comprehensive clinical data will be provided by the applicant in the future.
- Unmet medical needs will be fulfilled.
- The benefit to public health of the immediate availability of the medicine outweighs the risk that additional data are still required.
* ‘unmet medical needs’ means a condition for which there exists no satisfactory method of diagnosis, prevention or treatment authorised in the Community or, even if such a method exists, in relation to which the medicinal product concerned will be of major therapeutic advantage to those affected’ (Art. 4(2) of Regulation (EC) No 507/2006)
Conditions
Conditional marketing authorisations are valid for one year and can be renewed annually.
Once a conditional marketing authorisation has been granted, the marketing authorisation holder (MAH) must fulfil specific obligations within defined timelines. These obligations could include:
- completing ongoing or to conduct new studies or
- collecting additional data to confirm the medicine's benefit-risk balance remains positive
- specific obligations in relation to the collection of pharmacovigilance data
Of note: Specific obligations generally include clinical studies and exceptionally, in the context of public health emergencies, studies to provide further assurance on the pharmaceutical quality of e.g., vaccines.
The marketing authorisation can be converted into a standard marketing authorisation (no longer subject to specific obligations) once the marketing authorisation holder fulfils the obligations imposed and the complete data confirm that the medicine's benefits continue to outweigh its risks. Initially, this is valid for 5 years. It can then be renewed for unlimited validity.
As for any medicine, if new data show that the medicine’s benefits no longer outweigh its risks, EMA can take regulatory action, such as suspending or revoking the marketing authorisation. EMA can also take regulatory action if the company does not comply with the imposed obligations.
Report on 10 years of experience at the EMA avaliable at: Conditional marketing authorisation - Report on ten years of experience at the EMA (europa.eu)
Of note: A conditional marketing authorisation is different from an emergency use authorisation, which some countries use to permit the temporary use of an unauthorised medicine in an emergency situation. An emergency use authorisation is not a marketing authorisation.
Legal basis:
Article 14(7) of Regulation (EC) No 726/2004
Regulation (EC) No 507/2006
CHMP guideline on the scientific application and the practical arrangements necessary to implement Commission Regulation (EC) No 507/2006 on the conditional marketing authorisation for medicinal products for human use falling within the scope of Regulation (EC) No 726/2004.
1.1.1 Rolling review
A rolling review is a regulatory process that EMA uses to speed up the assessment of a promising medicine during a public health emergency. The EMA rolling review can accelerate every step of the regulatory pathway while ensuring that robust evidence on efficacy, safety and quality is generated to support scientific and regulatory decisions.
Normally, all data on a medicine or vaccine’s effectiveness, safety and quality and all required documents must be ready at the start of the evaluation for a formal marketing authorisation application (MAA). In the case of a rolling review, EMA’s CHMP reviews data as they become available from ongoing studies and can come to an opinion on the marketing authorisation sooner. Once the CHMP decides that sufficient data are available, the company can submit a formal MAA. EMA then has the option of recommending a conditional marketing authorisation. EU legislation foresees that conditional marketing authorisation is used as the fast-track authorisation procedure during public health emergencies to speed up approval and save lives.
A prime example for this approach is the Corona virus pandemic as described in the following.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the virus that causes COVID-19 (coronavirus disease 2019), the respiratory illness responsible for the COVID-19 pandemic. First identified in the city of Wuhan, Hubei, China, the World Health Organization declared the outbreak a Public Health Emergency of International Concern on 30 January 2020, and a pandemic on 11 March 2020. Epidemiological studies estimate that each infection results in an average of 2.39 to 3.44 new ones when no members of the community are immune and no preventive measures are taken.
The virus primarily spreads between people through close contact and via aerosols and respiratory droplets that are exhaled when talking, breathing, or otherwise exhaling, as well as those produced from coughs or sneezes. (adapted from WIKIPEDIA)
The EMA plays an important role in enabling the development, scientific evaluation, approval and monitoring of COVID-19 vaccines in the European Union (EU). EMA has adopted the rolling review procedure under its EMA emerging health threats plan as one measure to expedite the regulatory process and the availability of safe and efficacious vaccines against COVID-19.
The rolling review process in short:
- EMA's scientific committees – CHMP and Pharmacovigilance Risk Assessment Committee (PRAC)- review data as they become available on an ongoing basis.
- This review is done with the support of the COVID-19 EMA pandemic Task Force (COVID-ETF).
- The review takes place while development is still ongoing.
- The COVID-ETF has to agree on the start of a rolling review.
- There can be several rolling review cycles during the evaluation of one product while data continue to emerge. The number of cycles depends on the amount of data to be assessed.
- Each cycle is pre-agreed between EMA and the applicant.
- The submission for each cycle is done in eCTD (electronic Common Technical Document) format.
- In addition to the newly available data, each Rolling Review submission occurs in eCTD format with an application form, a CTD-Module 2 overview and responses to a cumulative listing of all outstanding questions from previous review cycles.
- During the rolling review, EMA assesses whether the data package is complete enough to invite the applicant to submit a formal marketing authorisation application.
End of rolling review - EMA processes the application under a shortened timetable.
- After review and upon positive recommendation by EMA the European Commission grants an EU-wide marketing authorisation
- National Competent Authorities decide on introduction of the newly approved vaccine and vaccination policies.
- Other data must be provided by the company, after approval (e.g., long-term protection data)
Figure 1, adapted from EMA, shows a graphic view of the rolling review process for COVID-19 vaccines:
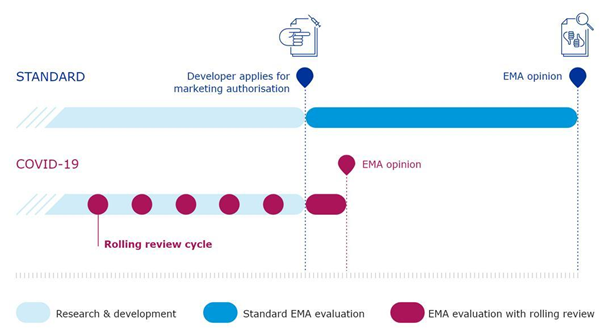
Figure 1: Graphic representation of the rolling review process for COVID-19 vaccines as deployed by EMA. The shortened time until the EMA opinion on the MAA is clearly visible.
Further reading: EMA initiatives for acceleration of development support and evaluation procedures for COVID-19 treatments and vaccines (16 September 2021 EMA/213341/2020 Rev.3)
EMA COVID-19 vaccines: development, evaluation, approval and monitoring
EMA COVID-19 guidance: evaluation and marketing authorisation