5. EU Regulatory procedures for a marketing authorisation (MA)
1. EU Regulatory procedures for a marketing authorisation (MA)
1.1. Centralised procedure
The EMA is responsible for the Centralised procedure (CP). A scientific evaluation of the submitted valid application is carried out by EMA’s Committee for Human Medicines (CHMP) and Pharmacovigilance Risk Assessment Committee (PRAC), the latter assessing the company’s proposed risk management plan (RMP). The CHMP adopts a final opinion with a recommendation to the EU Commission whether or not the medicine should be authorised at the end of the evaluation period which takes up to 210 days excluding additional time for clock stops. The EU Commission has the ultimate authority for granting the marketing authorisation within 67 days after receipt of the CHMP opinion. The MA is valid in the states of the European Economic Area (EEA) (all 27member states plus Liechtenstein, Norway and Iceland) under the same trade name (invented name). The great majority of new, innovative medicines pass through the centralised authorisation procedure.
Liechtenstein, Norway and Iceland) under the same trade name (invented name). The great majority of new, innovative medicines pass through the centralised authorisation procedure.
The CP is mandatory or optional for certain classes of products as shown below:
|
Mandatory for human medicines containing a new active substance to treat:
|
Optional for other human medicines:
|
*Regarding the criteria of ‘interest of patients’, a medicinal product which does not constitute a significant therapeutic, scientific or technical innovation, can be of patient interest at Union level when it addresses a specific health issue, allows access to medicines, or provides another type of contribution to patient care in the Union.
A brief overview of the centralised procedure, adapted from the EMA ( Obtaining an EU marketing authorisation, step-by-step and The evaluation of medicines, step-by-step), is given in the following table (Table 1) and flow chart (Fig. 4)
Table 1: Brief overview of temporal and operational sequence of the centralised procedure (CP) for marketing authorisations at the European Medicines Agency (EMA)
|
Months before submission |
Pre-submission – Actions |
|
18 to 7 7 7 to 6 3 to 2 |
Submission of eligibility request Notify EMA of the intended submission date (letter of intent) Pre-submission meetings (recommended) Re-confirmation of submission date - before submission Submission of the application – using the electronic common technical document (eCTD) format through the eSubmission gateway or web client Technical validation of the application by EMA to ensure all essential regulatory elements required for scientific assessment are included in the application prior to the start of the procedure. |
|
DAY |
Evaluation Procedure – ACTIONS |
|
1 |
Start of the procedure |
|
1 to 120 |
Preparation of assessment reports CHMP rapporteur’s and co-rapporteur’s teams assess the evidence provided on the medicine and independently prepare their assessment reports (AR) Inspection (if recommended) of the medicine’s manufacturing site Assessment of risk management plan rapporteur and co-rapporteur of EMA’s Pharmacovigilance Risk Assessment Committee (PRAC) assess the company’s proposed risk management plan (RMP), This assessment is reviewed by all PRAC members. List of questions ‘peer review’ meeting: discussion of comments from rapporteur and co-rapporteur teams, the other CHMP members, the PRAC members, and the CHMP peer reviewers, leading to a single AR and a list of concerns and objections. CHMP plenary meeting: discussion and adoption of the single AR. The report includes a final list of questions to be addressed by the applicant. |
|
120 |
*Clock stop 1 Applicant prepares responses to the CHMP’s questions and updates the RMP. Time frame: generally, three to six months. |
|
121 |
Restart of the clock |
|
121 to 180 |
Further assessment and list of outstanding issues Rapporteur and co-rapporteur evaluate the information sent by the applicant in response to the list of questions and update the AR which is reviewed and commented by the CHMP and PRAC members. The PRAC may at this stage request that the RMP include the conduct of safety studies after authorisation. Comments are consolidated and integrated into an updated AR which is discussed and adopted at a plenary meeting of the CHMP. Most of the time, this report will include a new list of questions, called the list of outstanding issues (LoOI). |
|
180 |
*Clock stop 2 Time frame: one to three months. |
|
181 |
Restart the clock An oral explanation can be requested either by applicant or CHMP. If so, the applicant is asked to provide clarifications on the committee’s outstanding issues. |
|
181 to 210 |
Further consultations EMA may consult external experts, e.g., calling on an EMA working party for specific questions or on additional experts, including patients and healthcare professionals, through a scientific advisory group or ad-hoc expert group meeting Final discussion and adoption of opinion After discussion as before, adoption of CHMP Opinion + CHMP Assessment Report:
|
|
215 at the latest |
Applicant provides the EMA with SmPC, labelling and PL in the official EU languages. EMA circulates draft translations to Member States for review. |
|
232 at the latest |
Applicant provides EMA with final translations of product information taking in account comments received from Member States by Day 229. |
|
By 237 |
Transmission of Opinion and Annexes in all EU languages to applicant, Commission, and Members of the Standing Committee, and Liechtenstein, Norway and Iceland. |
|
By 277 |
Within 67 days of receipt of CHMP opinion the European Commission either grants or refuses the marketing authorisation. Commission decisions are published in the Community Register of medicinal products for human use. EMA publishes a European public assessment report (EPAR) (introduce footnote explaining the different meanings AR, EPAR, PAR) for each medicine. When a new marketing authorisation application is refused, EMA publishes a refusal EPAR, including a question-and-answer document and an assessment report. |
*Clock stops relate to times allotted to the clarification of issues, where the process is interrupted for a specific period and resumes after the interruption with the previous time count.
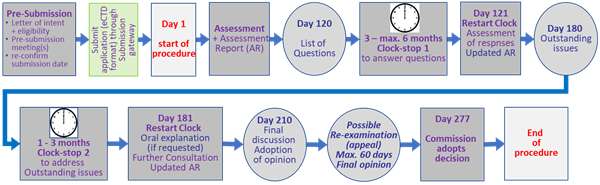
Figure 5: Flowchart of the centralised procedure (CP) for marketing authorisations at the European Medicines Agency (EMA
Legal basis: Regulation (EC) No 726/2004/EC