5. EU Regulatory procedures for a marketing authorisation (MA)
| Site: | EUPATI Open Classroom |
| Course: | Regulatory procedures- Marketing-Authorisations and their lifecycle management |
| Book: | 5. EU Regulatory procedures for a marketing authorisation (MA) |
| Printed by: | Guest user |
| Date: | Tuesday, 28 July 2026, 4:05 PM |
1. EU Regulatory procedures for a marketing authorisation (MA)
(This section is organised in the form of a book, please follow the blue arrows to navigate through the book or by following the navigation panel on the right side of the page.)
There are two principal ways for obtaining a MA:
- Centralised procedure (CP):
- Non-centralised procedures:
Each system has its own legal provisions and responsibilities for the competent authority (EMA or national competent authorities) and MA holders.
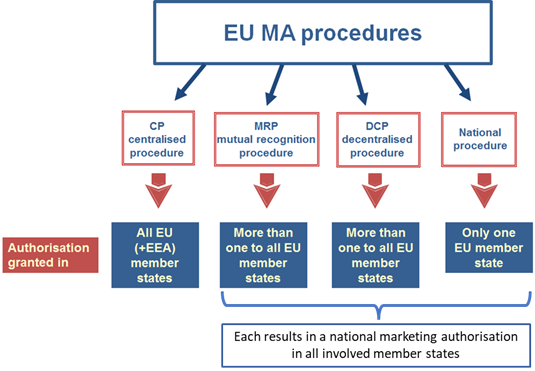
Figure 4: EU EMA procedure
Legal Basis CP: Regulation EC No.726/2004/EC
Legal Basis non-
CP: Directive 2001/83/EC as amended
1.1. Centralised procedure
The EMA is responsible for the Centralised procedure (CP). A scientific evaluation of the submitted valid application is carried out by EMA’s Committee for Human Medicines (CHMP) and Pharmacovigilance Risk Assessment Committee (PRAC), the latter assessing the company’s proposed risk management plan (RMP). The CHMP adopts a final opinion with a recommendation to the EU Commission whether or not the medicine should be authorised at the end of the evaluation period which takes up to 210 days excluding additional time for clock stops. The EU Commission has the ultimate authority for granting the marketing authorisation within 67 days after receipt of the CHMP opinion. The MA is valid in the states of the European Economic Area (EEA) (all 27member states plus Liechtenstein, Norway and Iceland) under the same trade name (invented name). The great majority of new, innovative medicines pass through the centralised authorisation procedure.
Liechtenstein, Norway and Iceland) under the same trade name (invented name). The great majority of new, innovative medicines pass through the centralised authorisation procedure.
The CP is mandatory or optional for certain classes of products as shown below:
|
Mandatory for human medicines containing a new active substance to treat:
|
Optional for other human medicines:
|
*Regarding the criteria of ‘interest of patients’, a medicinal product which does not constitute a significant therapeutic, scientific or technical innovation, can be of patient interest at Union level when it addresses a specific health issue, allows access to medicines, or provides another type of contribution to patient care in the Union.
A brief overview of the centralised procedure, adapted from the EMA ( Obtaining an EU marketing authorisation, step-by-step and The evaluation of medicines, step-by-step), is given in the following table (Table 1) and flow chart (Fig. 4)
Table 1: Brief overview of temporal and operational sequence of the centralised procedure (CP) for marketing authorisations at the European Medicines Agency (EMA)
|
Months before submission |
Pre-submission – Actions |
|
18 to 7 7 7 to 6 3 to 2 |
Submission of eligibility request Notify EMA of the intended submission date (letter of intent) Pre-submission meetings (recommended) Re-confirmation of submission date - before submission Submission of the application – using the electronic common technical document (eCTD) format through the eSubmission gateway or web client Technical validation of the application by EMA to ensure all essential regulatory elements required for scientific assessment are included in the application prior to the start of the procedure. |
|
DAY |
Evaluation Procedure – ACTIONS |
|
1 |
Start of the procedure |
|
1 to 120 |
Preparation of assessment reports CHMP rapporteur’s and co-rapporteur’s teams assess the evidence provided on the medicine and independently prepare their assessment reports (AR) Inspection (if recommended) of the medicine’s manufacturing site Assessment of risk management plan rapporteur and co-rapporteur of EMA’s Pharmacovigilance Risk Assessment Committee (PRAC) assess the company’s proposed risk management plan (RMP), This assessment is reviewed by all PRAC members. List of questions ‘peer review’ meeting: discussion of comments from rapporteur and co-rapporteur teams, the other CHMP members, the PRAC members, and the CHMP peer reviewers, leading to a single AR and a list of concerns and objections. CHMP plenary meeting: discussion and adoption of the single AR. The report includes a final list of questions to be addressed by the applicant. |
|
120 |
*Clock stop 1 Applicant prepares responses to the CHMP’s questions and updates the RMP. Time frame: generally, three to six months. |
|
121 |
Restart of the clock |
|
121 to 180 |
Further assessment and list of outstanding issues Rapporteur and co-rapporteur evaluate the information sent by the applicant in response to the list of questions and update the AR which is reviewed and commented by the CHMP and PRAC members. The PRAC may at this stage request that the RMP include the conduct of safety studies after authorisation. Comments are consolidated and integrated into an updated AR which is discussed and adopted at a plenary meeting of the CHMP. Most of the time, this report will include a new list of questions, called the list of outstanding issues (LoOI). |
|
180 |
*Clock stop 2 Time frame: one to three months. |
|
181 |
Restart the clock An oral explanation can be requested either by applicant or CHMP. If so, the applicant is asked to provide clarifications on the committee’s outstanding issues. |
|
181 to 210 |
Further consultations EMA may consult external experts, e.g., calling on an EMA working party for specific questions or on additional experts, including patients and healthcare professionals, through a scientific advisory group or ad-hoc expert group meeting Final discussion and adoption of opinion After discussion as before, adoption of CHMP Opinion + CHMP Assessment Report:
|
|
215 at the latest |
Applicant provides the EMA with SmPC, labelling and PL in the official EU languages. EMA circulates draft translations to Member States for review. |
|
232 at the latest |
Applicant provides EMA with final translations of product information taking in account comments received from Member States by Day 229. |
|
By 237 |
Transmission of Opinion and Annexes in all EU languages to applicant, Commission, and Members of the Standing Committee, and Liechtenstein, Norway and Iceland. |
|
By 277 |
Within 67 days of receipt of CHMP opinion the European Commission either grants or refuses the marketing authorisation. Commission decisions are published in the Community Register of medicinal products for human use. EMA publishes a European public assessment report (EPAR) (introduce footnote explaining the different meanings AR, EPAR, PAR) for each medicine. When a new marketing authorisation application is refused, EMA publishes a refusal EPAR, including a question-and-answer document and an assessment report. |
*Clock stops relate to times allotted to the clarification of issues, where the process is interrupted for a specific period and resumes after the interruption with the previous time count.
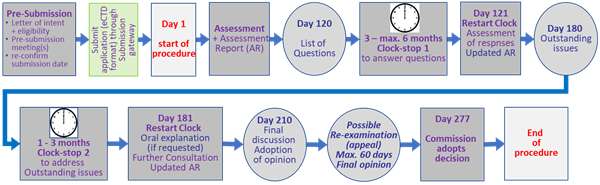
Figure 5: Flowchart of the centralised procedure (CP) for marketing authorisations at the European Medicines Agency (EMA
Legal basis: Regulation (EC) No 726/2004/EC
1.2. Non-centralised procedures
If a company wishes to request marketing authorisation (MA) for a medicine not eligible for or if the company does not opt for the centralised procedure it may use one of the following routes:
- National procedure (NP).
- Mutual recognition procedure (MRP), whereby a marketing authorisation granted in one Member State can be recognised in other EU countries
- Decentralised procedure (DCP), whereby a medicine that has not yet been authorised in the EU can be simultaneously authorised in several EU Member States.
To note: For all new MA applications in DCP, MRP and NP the eCTD format is mandatory. The data requirements and standards for the authorisation of medicines are the same in the EU, irrespective of the authorisation route.
1.3. Non-centralised procedures: 1. National procedure
The national procedures may be used under two circumstances:
1. Independent national procedures, strictly limited to medicines which are to be authorised and marketed in only one member state (MS).
If the medicine is intended to be marketed in more than one MS, the national procedure must be followed by a mutual recognition procedure. Alternatively, a decentralised procedure may be considered to obtain the MA in parallel in several MS.
2. For variations or extensions (additional strengths, pharmaceutical forms or routes of administration) of marketing authorisations already granted nationally (e.g., to harmonise the dossiers for subsequent use of the MRP or DCP).
MSs should ensure that the procedure for granting a MA is completed within a maximum of 210 days after the submission of a valid application. Clock stops are possible.
Legal basis: Artl. 8, 17, Directive 2001/83/EC; for variations: Artl. 4(1), 5(1) and 6(1) of Regulation (EC) No 1084/2003.
1.4. Non-centralised procedures:The Mutual recognition procedure and the decentralised procedure
Common characteristics
- The mutual recognition procedure or the decentralised procedure must be used for applications for marketing authorisation for medicines intended for marketing in more than one Member State
- Both procedures are based on the recognition by national competent authorities of an assessment performed by the authorities of one MS based on an identical dossier in these Member States.
- The procedures aim at facilitating access to a single market comprising the involved CMSs[1] by relying upon mutual recognition.
- It is not necessary to apply for a MA in all 27 MSs – a minimum of two is required.
- The application dossier is assessed by one member state (reference member state, RMS), the review is shared with other involved member states (concerned member state, CMS).
- The company has the choice of RMSs and CMS(s).
- The company has the choice of trade names.
- At the end of the procedure a national MA is issued by each member state involved, which includes a harmonised SmPC, labelling and Package leaflet (PL).
- Consideration should be given to the need for ‘user consultation’.
- In principle, CMSs should rely on the assessment of the RMS. Concerns may nevertheless be raised on grounds of a potential serious risks to public health. The issue, if not resolved between CMS, RMS and applicant, will be referred to the coordination group (CMD(h)). MSs shall reach a consensus within 60 days. In case this fails, the procedure is submitted to the EMA scientific committee (CHMP) for arbitration (Art. 29, Directive 2001/83/EC).
- Any application to vary a marketing authorization which has been granted under the MRP or DCP needs to be submitted to all the Member States which have previously authorised the medicine concerned (Art. 35 of Directive 2001/83/EC)
- The procedures are overseen by the Heads of Medicines Agencies (HMA) via the Coordination Group for Mutual Recognition and Decentralised Procedures - Human (CMD(h)).
1. Mutual recognition procedure - MRP
- The mutual recognition procedure is based on the principle of the mutual recognition by CMSs of a national MA where the medicine in question has received a MA in any Member State at the time of application.
- An identical application for mutual recognition is to be submitted to all concerned MSs and the applicant requests one Member State to act as RMS.
- The RMS either is asked to prepare an assessment report on the medicine or, if necessary, to update any existing assessment report.
- As soon as the AR is completed, copies of this report are sent to all CMSs, together with the approved SmPC, labelling and package leaflet.
- The concerned Member States shall recognise the marketing authorisation granted by the RMS.
- National marketing authorisations shall be granted within 30 days after acknowledgement of the agreement.
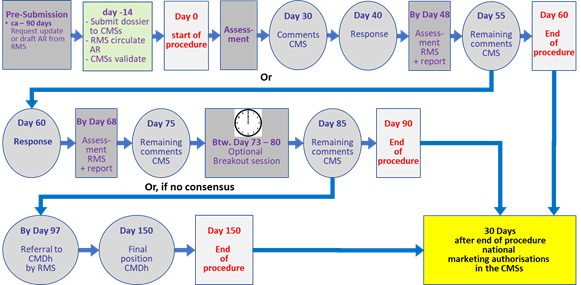
The following table (Table 2) and flowchart (Fig.5) give an overview and timeline of the MRP.
Table 2: Brief overview of temporal and operational sequence of the mutual recognition procedure (MRP) for marketing authorisations in more than one member state (MS).
|
Approx. 90 days before submission to CMS(s) |
Applicant requests RMS to update Assessment Report (AR) and allocate procedure number. |
|
14 days before start of procedure |
Applicant submits dossier to CMS(s). RMS circulates AR including SmPC, PL and labelling to CMSs. Validation of the application by CMSs. |
|
Day 0 |
RMS starts procedure |
|
Day 30 |
CMSs send their comments to the RMS, CMSs and applicant |
|
Day 40 |
Applicant sends the response document to CMSs and RMS |
|
Until Day 48 |
RMS evaluates and circulates a report on the applicant’s response document to CMSs. |
|
Day 55 |
CMSs send their remaining comments to RMS and applicant. |
|
Day 55-59 |
The applicant and RMS are in close contact to clarify if the procedure can be closed at day 60 or if the applicant should submit a further response at day 60. |
|
Day 60 |
If CMS have no remaining comments at Day 55, the RMS closes the procedure. If a CMS has remaining comments at Day 55, the applicant sends the response document to CMSs and RMS |
|
Day 60 - 90 |
Only used if a CMS has remaining comments |
|
Until day 68 |
RMS evaluates and circulates a report on the applicant’s response document to CMSs |
|
Day 75 |
CMSs send their remaining comments to RMS, CMSs and applicant. |
|
Until Day 80 |
Optional break-out session (BOS) can be organised around Day 75 (but may take place between days 73-80) |
|
Day 85 |
CMSs send any remaining comments to RMS, CMS and applicant |
|
Day 90 |
CMSs notify RMS and applicant of final position (in case of negative position also the CMDh secretariat of EMA). If consensus is reached, the RMS closes the procedure.
If consensus is not reached, the points for disagreement submitted by CMSs are referred to CMDh by the RMS within 7 days after day 90 |
|
Day 150 |
Final position adopted by the CMDh: If consensus is reached at the level of CMDh, the RMS closes the procedure. If consensus is not reached at the level of CMDh, the RMS refers immediately the matter to EMA for CHMP arbitration |
|
7 days after close of procedure |
Applicant sends high quality national translations of SmPC, PL and labelling to CMSs. |
|
30 days after close of procedure |
Granting of national marketing authorisations in the CMSs subject to submission of acceptable translations |
- The DCP is available for new products in cases where the medicine has no marketing authorisation at the time of application.
- It is a single, streamlined procedure with the possibility for shortened approval times in straightforward cases.
- It is possible to end the procedure at any time point during the procedure if consensus is reached.
- Submission of an application in all the MSs involved at the same time.
- One MS acts as the RMS and takes the lead (this can be suggested by the sponsor)
- The RMS prepares a draft assessment report, including SmPC, labelling and PL and sends them to the CMSs and to the applicant.
- The CMSs shall approve the assessment report, the SmPC and the labelling and PL.
- National MAs will be granted within 30 days after acknowledgement of the agreement in the RMS and each CMS with a harmonised SmPC, labelling and PL
The following table (Table 3) and flowchart (Fig. 6) give an overview and timeline of the DCP.
Table 3: Brief overview of temporal and operational sequence of the decentralised procedure (DCP) for marketing authorisations in more than one member state (MS)
|
Pre-procedural Step |
|
|
14 days before submission |
Applicant discussions with RMS. RMS allocates procedure number |
|
14 days before start of procedure |
Submission of the dossier to the RMS and CMSs. Validation of the application. |
|
Assessment step I |
|
|
Day 0 |
RMS starts the procedure. CMSs are informed |
|
Day 70 |
RMS forwards the Preliminary Assessment Report (PrAR) (including comments on SmPC, PL and labelling) to the CMSs and the applicant. |
|
Until Day 100 |
CMSs send their comments, if there are any, to the RMS, CMSs and applicant |
|
Until Day 105 |
Consultation between RMS and CMSs and applicant. If consensus not reached RMS stops the clock to allow applicant to supplement the dossier and respond to the questions. |
|
Clock-stop period |
Applicant sends final response document to the RMS and CMSs within a period of 3 months, which can be extended by a further 3 months. |
|
Day 106 |
RMS restarts the procedure after receipt of a final response or expiry of the agreed clock-stop period without a response. CMSs are informed |
|
Assessment step II |
|
|
Day 120 (Day 0) |
RMS sends the draft AR, including SmPC, labelling and PL to CMSs and the applicant. |
|
Day 145 (Day 25) |
CMSs send comments, if there are any, to RMS, CMSs and the applicant. |
|
Day 150 (Day 30) |
RMS may close procedure if consensus reached. CMSs then have 30 days to grant the MA |
|
Day 160 |
Applicant sends the response document to CMSs and RMS. |
|
Until 180 (Day 60) |
If consensus is not reached by day 150, RMS to communicate outstanding issues with applicant, receive any additional clarification, prepare a short report and forward it to the CMSs and the applicant. |
|
Day 195 (at the latest) |
A Break-Out Session (BOS) may be held at the EMA (or via TC) with the involved MSs to reach consensus on the major outstanding issues. |
|
Between Day 195 and Day 210 |
RMS consults with the CMSs and the applicant to discuss the remaining comments raised. |
|
Day 210 (Day 90) |
If consensus is reached: - In case of positive position from RMS, Closure of the procedure including End of Procedure letter, final Day 210 overview AR, SmPC, labelling and PL, active substance and finished product specifications and proceed to national 30 days step for granting the MA. - In case of negative position from the RMS, closure of the procedure negatively. The End of Procedure letter and final Day 210 overview AR is circulated. If consensus is not reached: In case of negative position from CMS, CMS notifies the RMS, the other CMSs, applicant and the secretariat of the CMDh. Referral to the CMDh. |
|
At the latest, within 7 days after Day 210 |
If consensus on a positive RMS AR was not reached at day 210, the points of disagreement submitted by CMS(s) will be referred by the RMS to the CMDh for resolution. |
|
Day 270 (at the latest) |
Final position adopted by CMDh with referral to CHMP for arbitration in case of unsolved disagreement. |
* The Communication and Tracking System (CTS) is the system used by the National Competent Authorities (NCAs) involved in the licensing of human and veterinary medicinal products via the MRP and DCP. CTS supports the co-ordination and tracking of marketing authorisation, post-licensing and work sharing procedures as monitored by the Coordination Groups for MRP and DCP. The system serves as data provider for other applications.
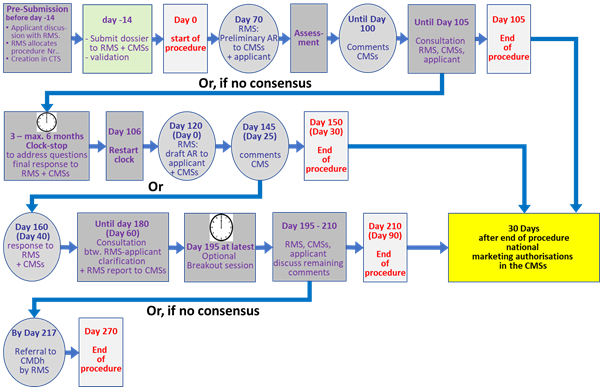
Figure 7: Flowchart of the decentralised procedure (DCP)
Legal basis for MRP and DCP: Artl. 8 (1), (3); 17; 10, 10a,b,c; 11; 18; 28 to 39, Directive 2001/83 (EC)
[1] A single market is referring to a multitude of entities still being considered as ‘one’. For example, the EU market is a single market.
1.5. Non-centralised procedures: Objections to mutual recognition in MRP and DCP
Objections to recognition in the MRP and DCP can only be raised on the grounds of potential serious risk to public health. The issue, if not resolved between CMS, RMS and applicant, will be referred to the coordination group (CMD(h)) where within 60 days, MSs shall reach a consensus. In case this fails, the procedure is submitted to the EMA scientific committee (CHMP) for arbitration.
In the case that at least one of the concerned Member States objects to approve the assessment report or acknowledge the MA of the RMS on the grounds of a potential serious risk to public health, it shall give a detailed explanation of the reasons for its position to the RMS, to the other CMSs and to the applicant.
Based on Article 29 (2) of Directive 2001/83/EC as amended, a guideline has been adopted to set out in more detail in which exceptional cases a Member State concerned in a mutual recognition procedure or in a decentralised procedure can refuse to recognise a marketing authorisation or a positive assessment on the basis of a potential serious risk to public health.
In the following, this guideline (adapted)will be used to describe what can be considered a serious risk to public health and therefore would constitute the basis for a concerned Member State to raise major objections and to establish clearly and substantiated why the proposed authorisation (or refusal) should not be accepted.
Excerpt from: ‘Guideline on the definition of a potential serious risk to public health in the context of Article 29 (1) and (2) of Directive 2001/83/EC — March 2006 (2006/C 133/05):
Directive 2001/83/EC does not provide for a definition of a ‘potential serious risk to public health’. However, the following definitions are given in that Directive:
- The term ‘risk related to the use of the medicine’ is defined in point 28 of Article 1, first indent of Directive 2001/83/EC as ‘any risk relating to the quality, safety or efficacy of the medicine as regards to patients' health or public health’ (or any risk of undesirable effects on the environment).
- The term ‘risk-benefit balance’ is defined in point 28a of Article 1 of that Directive as ‘an evaluation of the positive therapeutic effects of the medicine in relation to the risks as defined in point 28, first indent’.
For the application of this guideline, the following definitions shall apply:
- A ‘risk’ is defined as the probability that an event will occur.
- A ‘potential serious risk to public health’ is defined as a situation where there is a significant probability that a serious hazard resulting from a human medicine in the context of its proposed use will affect public health.
- ‘Serious’ in this context means a hazard that could result in death, could be life-threatening, could result in patient hospitalisation or prolongation of existing hospitalisation, could result in persistent or significant disability or incapacity, or could be a congenital anomaly/birth defect or permanent or prolonged signs in exposed humans.
1. Potential serious risk to public health
The assessment of a potential serious risk to public health cannot be made in isolation but has to take into account the positive therapeutic effects of the medicine in question. Consequently, the term potential serious risk to public health as used in Article 29(1) of Directive 2001/83/EC has to be understood as relating to the overall benefit-risk assessment of the medicine.
Any major objection must be scientifically justified and consider the nature and degree of any hazards, the magnitude of the risks involved, the benefits associated with the use of the product and the feasibility and practicality of the implementation of any measures to mitigate the risks.
A potential serious risk to public health in relation to a particular medicine may mainly exist under the following circumstances:
1. Efficacy:
-
- The data submitted to support therapeutic efficacy do not provide sound scientific justification for the claims for efficacy.
- Adequate proof for bioequivalence demonstrated by generic medicines to the reference medicine is lacking.
2. Safety:
-
- The evaluation of the non-clinical and clinical safety data and post-marketing data does not provide adequate support for the conclusion that all potential safety issues for the target population have been appropriately and adequately addressed in the proposed labelling.
- The absolute level of risk from the medicine, in the context of its proposed use, is considered unacceptable.
3. Quality:
-
- The proposed production and quality control methods cannot guarantee that a major deficiency in the quality of the product will not occur.
-
- The benefit-risk balance for the product is not considered to be favourable, taking into account the nature of the identified risk (s) and the potential benefit in the proposed indication (s) and target patient population (s).
5. Product Information:
-
- The information is misleading or incorrect for either the prescribers or the patients to ensure the safe use of the medicine.
Any objection on the ground of a potential serious risk to public health cannot be justified by differences in national administrative or national scientific requirements, or internal national policies, unless the conditions above or Article 29(1) of Directive 2001/83/EC are fulfilled.