2. Summary of Product Characteristics (SmPC)
1. Summary of Product Characteristics (SmPC)
1.1. SmPC process in an application for marketing authorisation
Establishing the SmPC is an essential part of the approval process for a new medicine.
The pharmaceutical company proposes the text of the SmPC in the dossier. In addition to the requirements described in the EU directive, a Commission Guideline on the SmPC is available to industry. The SmPC proposal is included in Module 1 (CTD) in the application (CTD: The Common Technical Document, organized into five modules. Module 1 is region specific and modules 2, 3, 4 and 5 are common for all regions). The assessors evaluating the application dossier will consider if the information of the proposed SmPC is correct and contains the required items. The SmPC therefore is the result of the agreed position on the medicinal product, as distilled during the assessment process. The SmPC forms an integral part of the marketing authorisation (MA). Therefore, the company cannot change the SmPC after approval without submitting a so-called variation application to the regulatory authorities who may also initiate and impose changes, for example, if new adverse reactions (ADR) occur. The following figure (Fig. 1) is a simplified schematic of the process of SmPC authoring, approval and management.
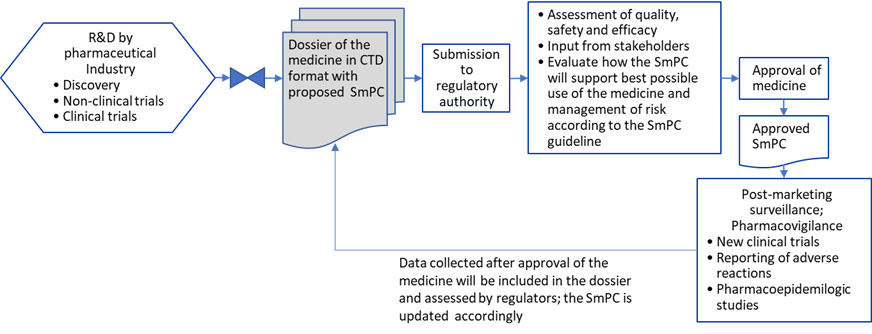
Figure.1: Schematic representation of the SmPC process
The European Medicines Agency's (EMA) Working Group on Quality Review of Documents (QRD) develops, reviews and updates templates for product information for use by applicants and marketing authorisation holders for human medicines. The template can be found here: (https://www.ema.europa.eu/en/documents/template-form/qrd-product-information-annotated-template-english-version-102-rev1_en.pdf).
This practical advice on how to draw up the SmPC sets out content and order of the required information, including guidance and explanatory notes as well as a reference to an additional guidance detailing the conventions to be followed when using the template. Find out how to prepare and review a summary of product characteristics
Information in the SmPC should be presented according to a predefined structure. Some information may be suitable in different sections, but cross-references are made to avoid repetitive information.
For the interested reader, please see in the box below the introductory note of the QRD template.
ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
[NOTE: the following are those items of information required by Article 11 of Directive 2001/83/EC and current practice in the centralised procedure. In the case of advanced therapy medicinal products, these items are listed in Annex II of Regulation (EC) 1394/2007.
For the full information to be included in each section, please refer to the “Guideline on Summary of Product Characteristics” as published on the website of the European Commission in the Notice to Applicants, Volume 2C: https://ec.europa.eu/health/system/files/2016-11/smpc_guideline_rev2_en_0.pdf
This guidance should also be read in conjunction with other relevant guidelines that can be found on the European Medicines Agency website (e.g. “QRD Convention to be followed for the EMA-QRD templates”
The use of combined SmPCs for different strengths…
See also: “The Product Information linguistic review process for new applications in the Centralised Procedure”: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/linguistic-review-process-product-information-centralised-procedure-human_en.pdf
Standard statements are given in the template, which must be used whenever they are applicable. If the applicant needs to deviate from these statements to accommodate medicinal product-specific requirements, alternative or additional statements will be considered on a case-by-case basis.