2. Summary of Product Characteristics (SmPC)
| Site: | EUPATI Open Classroom |
| Course: | Product information and information to the public |
| Book: | 2. Summary of Product Characteristics (SmPC) |
| Printed by: | Guest user |
| Date: | Friday, 7 August 2026, 12:17 AM |
1. Summary of Product Characteristics (SmPC)
(This section is organised in the form of a book, please follow the blue arrows to navigate through the book or by following the navigation panel on the right side of the page.)
SmPCs are an integral part of the marketing authorisation of all medicines authorised in the European Union and the basis of information for healthcare professionals on how to use a medicine safely and effectively. They are kept updated throughout the lifecycle of a medicine as new efficacy or safety data emerge.

SmPCs are also the basis for the preparation of package leaflets, and therefore are important documents in enabling information on medicines to reach patients.
As regards the legal background, the primary legislation is Directive 2001/83/EC which stipulates:
- The application shall be accompanied by the following particulars and documents, submitted in accordance with Annex I:… A summary, in accordance with Article 11, of the product characteristics (Art. 8 (3)(j))
- The summary of the product characteristics shall contain, in the order indicated below, the following information: [it follows the list of items to be included] (Art. 11)
- ANNEX I Summary of Product Characteristics [NOTE: the following are those items of information required by Article 11 of Directive 2001/83/EC] Version 10.2 rev. 1, 02/2021 (see next).
1.1. SmPC process in an application for marketing authorisation
Establishing the SmPC is an essential part of the approval process for a new medicine.
The pharmaceutical company proposes the text of the SmPC in the dossier. In addition to the requirements described in the EU directive, a Commission Guideline on the SmPC is available to industry. The SmPC proposal is included in Module 1 (CTD) in the application (CTD: The Common Technical Document, organized into five modules. Module 1 is region specific and modules 2, 3, 4 and 5 are common for all regions). The assessors evaluating the application dossier will consider if the information of the proposed SmPC is correct and contains the required items. The SmPC therefore is the result of the agreed position on the medicinal product, as distilled during the assessment process. The SmPC forms an integral part of the marketing authorisation (MA). Therefore, the company cannot change the SmPC after approval without submitting a so-called variation application to the regulatory authorities who may also initiate and impose changes, for example, if new adverse reactions (ADR) occur. The following figure (Fig. 1) is a simplified schematic of the process of SmPC authoring, approval and management.
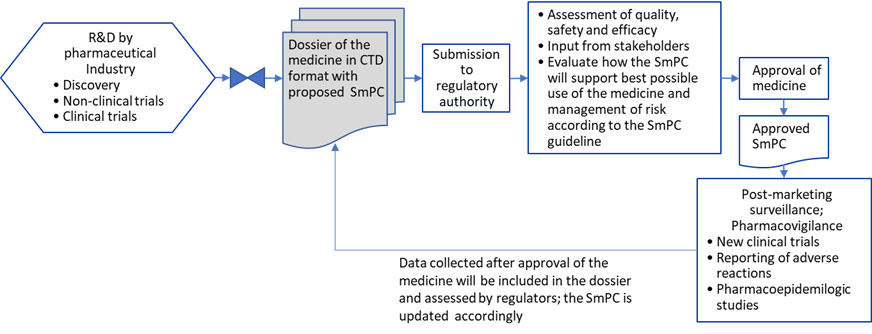
Figure.1: Schematic representation of the SmPC process
The European Medicines Agency's (EMA) Working Group on Quality Review of Documents (QRD) develops, reviews and updates templates for product information for use by applicants and marketing authorisation holders for human medicines. The template can be found here: (https://www.ema.europa.eu/en/documents/template-form/qrd-product-information-annotated-template-english-version-102-rev1_en.pdf).
This practical advice on how to draw up the SmPC sets out content and order of the required information, including guidance and explanatory notes as well as a reference to an additional guidance detailing the conventions to be followed when using the template. Find out how to prepare and review a summary of product characteristics
Information in the SmPC should be presented according to a predefined structure. Some information may be suitable in different sections, but cross-references are made to avoid repetitive information.
For the interested reader, please see in the box below the introductory note of the QRD template.
ANNEX I
SUMMARY OF PRODUCT CHARACTERISTICS
[NOTE: the following are those items of information required by Article 11 of Directive 2001/83/EC and current practice in the centralised procedure. In the case of advanced therapy medicinal products, these items are listed in Annex II of Regulation (EC) 1394/2007.
For the full information to be included in each section, please refer to the “Guideline on Summary of Product Characteristics” as published on the website of the European Commission in the Notice to Applicants, Volume 2C: https://ec.europa.eu/health/system/files/2016-11/smpc_guideline_rev2_en_0.pdf
This guidance should also be read in conjunction with other relevant guidelines that can be found on the European Medicines Agency website (e.g. “QRD Convention to be followed for the EMA-QRD templates”
The use of combined SmPCs for different strengths…
See also: “The Product Information linguistic review process for new applications in the Centralised Procedure”: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/linguistic-review-process-product-information-centralised-procedure-human_en.pdf
Standard statements are given in the template, which must be used whenever they are applicable. If the applicant needs to deviate from these statements to accommodate medicinal product-specific requirements, alternative or additional statements will be considered on a case-by-case basis.
1.2. Structure and content of the SmPC
Quotes from the template are included to illustrate exemplary the complexity and detail of the SmPC, incl. prescriptive text. These quotes are indicated in the following way: [Green text]. They are guidance and explanatory notes only, to be deleted when using the templates. Additional explanatory notes (not in the QRD template) are inserted directly after the respective heading in brackets and ‘italic’
{text}: Information to be filled in
<text>: Text to be selected or deleted as appropriate
The black symbol and the statements should only appear preceding section 1. The black symbol shall be a black inverted equilateral triangle: the symbol shall be proportional to the font size of the subsequent standardized text and in any case each side of the triangle shall have a minimum length of 5 mm. For the purpose of preparing the product information annexes please use the black triangle as presented in this template (see below).]
<  This medicinal product is subject to additional monitoring. This will allow quick identification of
This medicinal product is subject to additional monitoring. This will allow quick identification of
new safety information. Healthcare professionals are asked to report any suspected adverse reactions. <See section 4.8 for how to report adverse reactions.>
1. Name of the medicinal product, strength and pharmaceutical form.
2.. Qualitative and quantitative composition.
3. Pharmaceutical form.
4. Clinical particulars.
4.1. Therapeutic indication(s) (define unambiguously for whom the medicine is indicated specifying any limitation, e.g. age)
4.2. Posology[i] and method of administration (clearly specify the posology for each indication and relevant patient category for which the medicine is indicated, including any special population in which dose adjustment may be required)
4.3. Contraindications
4.4. Special warnings and precautions for use
4.5. Interaction with other medicinal products and other forms of interaction (highlight the clinically relevant interactions which result in a recommendation for use)
4.6. Fertility, pregnancy and lactation (Follow the list of pregnancy and lactation statements, in an (Appendix I).)
4.7. Effects on ability to drive and use machines
4.8. Undesirable effects (summarise the safety profile of the medicine and list all adverse reactions (but not the adverse events))
<Paediatric population>
[For ALL medicinal products: The following sub-heading should appear at the end of section 4.8:]
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.*
[*For the printed materials: No reference to Appendix V should be included in the printed materials….]
4.9. Overdose
5. Pharmacological properties. (This includes information about how the medicine works, safety information, how the medicine is absorbed and removed from the body.)
5..1. Pharmacodynamic properties (may provide limited information on clinical results if it is relevant to the prescriber, statistically compelling and supports the authorised indication(s))
[For medicinal products authorised as similar biological medicinal products, include the following statement:]
<{(Invented) name} is a biosimilar medicinal product. Detailed information is available on the website
of the European Medicines Agency http://www.ema.europa.eu.>
5.2. Pharmacokinetic properties
<Absorption>
<Distribution>
<Biotransformation>
<Elimination>
<Linearity/non-linearity>
[Additional sub-heading(s), such as “Renal impairment”, “Hepatic impairment”, “Elderly”, “Paediatric population” or “Other special populations” (to be specified) should be used, where appropriate.]
<Pharmacokinetic/pharmacodynamic relationship(s)>
5. 3. Preclinical safety data
[Additional sub-headings such as “Juvenile animal studies” can be included when necessary.]
<Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, toxicity to reproduction and development.>
<Effects in non-clinical studies were observed only at exposures considered sufficiently in excess of the maximum human exposure indicating little relevance to clinical use.>
<Adverse reactions not observed in clinical studies, but seen in animals at exposure levels similar to clinical exposure levels and with possible relevance to clinical use were as follows:>
<Environmental risk assessment (ERA)>
6. Pharmaceutical particulars.
6.1. List of excipients
6.2. Incompatibilities
6.3. Shelf life
6.4. Special precautions for storage
[For storage condition statements, see Appendix III.]
[General storage conditions of the finished medicinal product should appear here, together with a cross-reference to section 6.3 where appropriate:]
<For storage conditions after <reconstitution><dilution><first opening> of the medicinal product, see section 6.3.>
6.5. Nature and contents of container <and special equipment for use, administration or implantation>
[The proposed optional heading “and special equipment for use, administration or implantation” is for advanced therapy medicinal products only. Explanatory illustrations may be included, if necessary.]
[Multipack presentations should also be listed in this section, e.g. “multipacks containing 180 (2 packs of 90) film-coated tablets”.]
<Not all pack sizes may be marketed.>
6.6. Special precautions for disposal <and other handling>
7. Marketing authorisation holder.
8. Marketing authorisation number(s).
9. Date of first authorisation/renewal of the authorisation.
10. Date of revision of the text.
For detailed information on a medicinal product, reference should be made to the public assessment report within the SmPC (e.g. “Detailed information on this product is available on the website of the European Medicines Agency (EMEA) http://www.ema.europa.eu”)
When planning the treatment of a patient, the SmPC is a useful tool for the doctor. It contains all information the doctor needs to inform the patient on the benefits and the risks of the chosen medicine. Furthermore, guidance on the use is included along with relevant warnings on what to do and what to avoid during the treatment.