What is pharmacovigilance (PhV)?
5. The PhV process in short
The entire PhV process—from systems that monitor for and detect possible adverse effects (safety signals) through to regulatory action to mitigate risks—is coordinated across the regulatory network, the pharmaceutical industry and health systems. The PhV system receives a wide range of input including that from non-EU regulators, academia, health professionals and patients and via several other channels.
A signal is defined as “information arising from one or multiple sources, including observations and experiments, which suggest a new potentially causal association, or a new aspect of a known association between an intervention and an event or set of related events, either adverse or beneficial, that is judged to be of sufficient likelihood to justify verificatory action.”
(Commission Implementing Regulation (EU) No. 520/2012 of 19 June 2012 on the performance of pharmacovigilance activities provided for in Regulation (EC) No. 726/2004 of the European Parliament and of the Council and Directive 2001/83/ EC of the European Parliament and of the Council. EUROPEAN COMMISSION https://eur-lex.europa.eu/eli/reg_impl/2012/520/oj
Signal management in the EU focuses on adverse outcomes and involves a set of activities performed to determine whether there are new risks associated with an active substance or a medicinal product or whether risks have changed.
(Guideline on good pharmacovigilance practices (GVP): module IX: signal management (Rev 1). EUROPEAN MEDICINES AGENCY <https://www.ema.europa.eu/en/documents/scientific-guideline/ guideline-good-pharmacovigilance-practices-gvp-module-ix-sig-nal-management-rev-1_en.pdf> (2017).)
A safety signal therefore is information on a new or known adverse event that is potentially caused by a medicine and that warrants further investigation.
(COMMISSION STAFF WORKING DOCUMENT (2017) https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52017SC0201&from=EN)
In the following a brief outline of the PhV process is given including the concepts, the steps followed and the actors involved when dealing with a suspected adverse reaction of a medicine (how to come from an adverse event to an adverse reaction (ADR) (see below).
The process starts with the PhV system receiving information from which a signal for a suspected adverse reaction of a medicine is identified (signal detection, performed by the EMA, Member States and MAHs). For centrally authorised medicinal products (CAPs), the EMA is responsible for EudraVigilance data monitoring in collaboration with PRAC, and Member States, in collaboration with the EMA for medicinal products authorised nationally (NAPs). Figure 3 shows the process steps together with explanatory notes in a high-level schematic.
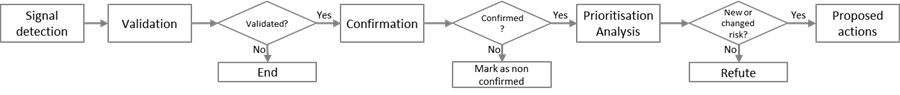
Figure.3: High-level workflow schematic of the pharmacovigilance signal management process. Adapted from: HMA Guideline on good pharmacovigilance practices (GVP)Module IX – Signal management (Rev 1) (2017) available at https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-gvp-module-ix-signal-management-rev-1_en.pdf
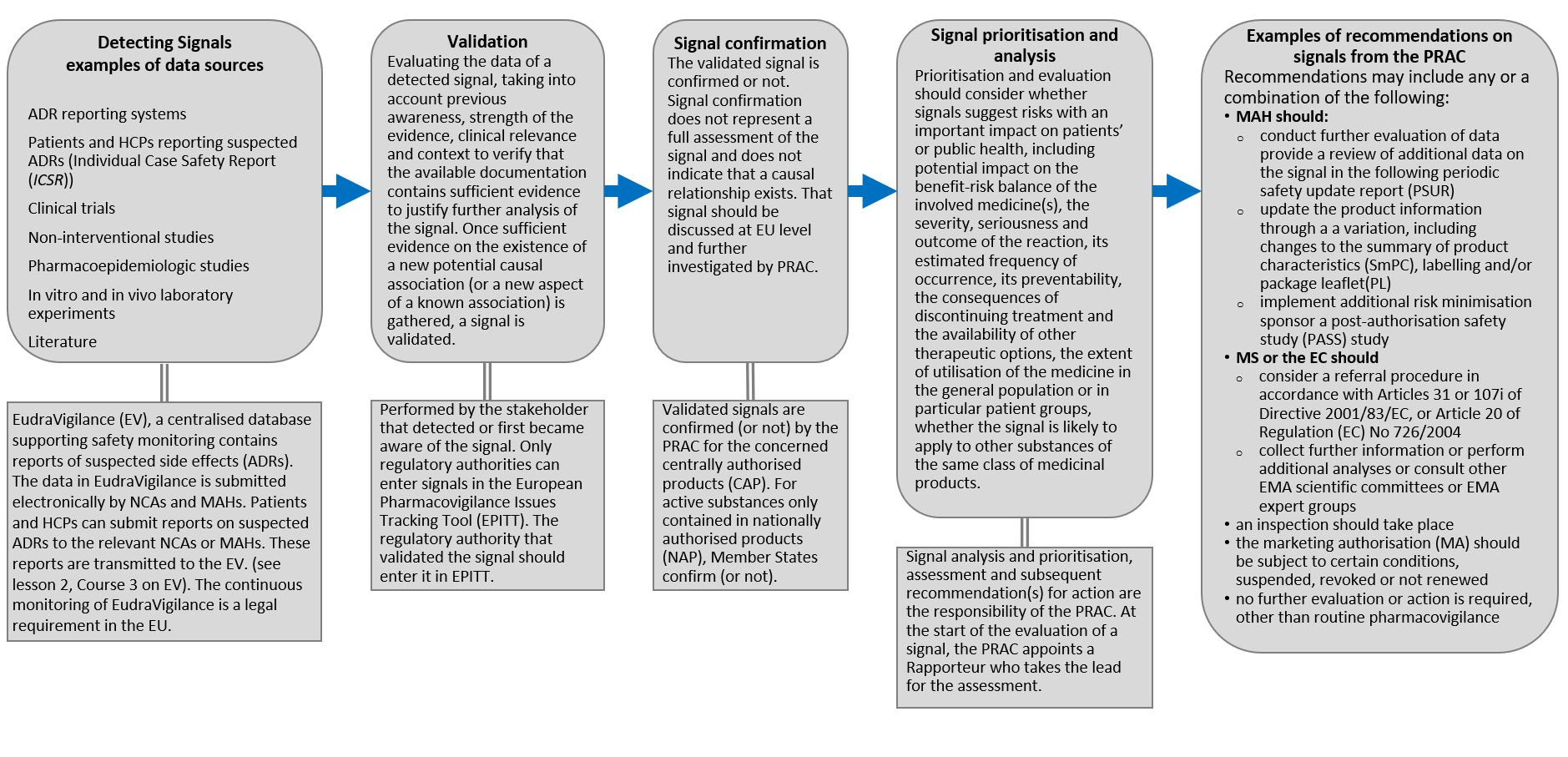
Figure.4: High-level workflow schematic of the pharmacovigilance signal management process. Adapted from: HMA Guideline on good pharmacovigilance practices (GVP)Module IX – Signal management (Rev 1) (2017) available at https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-gvp-module-ix-signal-management-rev-1_en.pdf
The following gives a short overview of the steps in the signal management process.
Detecting Signals examples of data sources: ADR reporting systems, Patients and HCPs reporting suspected ADRs (Individual Case Safety Report (ICSR)), Clinical trials, Non-interventional studies, Pharmacoepidemiologic studies, In vitro and in vivo laboratory experiments, Literature etc.
Signal data are transmitted and held in the EudraVigilance (EV), a centralised database supporting safety monitoring that contains reports of suspected adverse reactions (ADRs). The data in EudraVigilance is submitted electronically by NCAs and MAHs. Patients and HCPs can submit reports on suspected ADRs to the relevant NCAs or MAHs. The continuous monitoring of EudraVigilance is a legal requirement in the EU (see lesson 2 on EV).
Validation: Evaluating the data of a detected signal, taking into account previous awareness, strength of the evidence, clinical relevance and context to verify that the available documentation contains sufficient evidence to justify further analysis of the signal. Once sufficient evidence on the existence of a new potential causal association (or a new aspect of a known association) is gathered, a signal is validated. Performed by the stakeholder that detected or first became aware of the signal. Only regulatory authorities can enter signals in the European Pharmacovigilance Issues Tracking Tool (EPITT). The regulatory authority that validated the signal should enter it in EPITT.
Signal confirmation: The validated signal is confirmed or not. Signal confirmation does not represent a full assessment of the signal and does not indicate that a causal relationship exists. That signal should be discussed at EU level and further investigated by PRAC. Validated signals are confirmed (or not) by the PRAC for the concerned centrally authorised product (CAP). For active substances only contained in nationally authorised products (NAP), Member States confirm (or not).
Signal prioritisation and analysis: should consider
- whether signals suggest risks with an important impact on patients’ or public health, including potential impact on the benefit-risk balance of the involved medicine(s),
- the severity, seriousness and outcome of the reaction, its estimated frequency of occurrence, its preventability, the consequences of discontinuing treatment and the availability of other therapeutic options,
- the extent of utilisation of the medicine in the general population or in particular patient groups,
- whether the signal is likely to apply to other substances of the same class of medicinal products.
Signal analysis and prioritisation, assessment and subsequent recommendation(s) for action are the responsibility of the PRAC. At the start of the evaluation of a signal, the PRAC appoints a Rapporteur who takes the lead for the assessment.
Examples of recommendations on signals from the PRAC: Recommendations may include any or a combination of the following:
- MAH should:
- conduct further evaluation of data and provide a review of additional data on the signal in the following periodic safety update report (PSUR
- update the product information through a variation, including changes to the summary of product characteristics (SmPC), labelling and/or package leaflet (PL)
- implement additional risk minimisation measures
- sponsor a post-authorisation safety study (PASS) study
- MS or the EC should
- an inspection should take place
- the marketing authorisation (MA) should be subject to certain conditions, suspended, revoked or not renewed
-
no further evaluation or action is required, other than routine pharmacovigilance
Adapted from
- https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/questions-answers-article-31-pharmacovigilance-referral-procedures_en.pdf
- https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/questions-answers-urgent-union-procedures-article-107i-directive-2001/83/ec_en.pdf
- https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-gvp-module-ix-signal-management-rev-1_en.pdf