1. Introduction: Regulatory affairs in the medicines development process
Introduction: Regulatory affairs in the medicines development process
The medicines development process is a long journey in a number of defined steps, with various decision points along the way as shown in Figure 1. (For more details see also Course 1). The goal for any development process is ultimately an approval for marketing of the new medicine and access for patients.

Figure 1: Overview of development steps, decision points, timelines in medicines R&D. Red arrow indicates theme of this lesson.
The phase marked ‘Regulatory Review’ in Fig. 1 and section ‘Regulatory approval’ is the theme of this lesson. It covers the EU legal background and regulatory procedures for Marketing Authorisation (MA). This is also the development step where the Regulatory Affairs (RA) function, which is an integral and important part of all development steps, is most active.
The RA is also involved with the lifecycle management of a medicine. Lifecycle management include modifications that are planned or required post-authorisation, such as product variations or additional studies.
RA provides the regulatory strategy and expertise which encompass, but are not limited to:
- regulatory objectives
- regional/national regulatory requirements, hurdles
- the regulatory landscape and precedents and competition
- risks to potential success in delivering a specific regulatory outcome
- regulatory intelligence, including national/local expectations
- marketing authorisation options and their advantages/drawbacks
- correspondence and/or contacts with respective regulatory authorities
- document management, including dossier compilation for Marketing Authorisation Application (MAA)
- regulatory submission management, including clinical trial applications (CTAs)
- regulatory input to advertising and promotion
- provision of specific functional expertise such as labelling, CMC, nonclinical and clinical.
A global regulatory strategy ideally combines regulatory and business objectives because regulatory decisions can affect the time-to-market, label claims and reimbursement and, consequently, likely business outcomes. Equally, consideration should be given to broader implications such as links to health outcomes, the potential for patient access, and/or effects on the healthcare system (e.g., cost effectiveness). This should be considered particularly in light of the Health Technology Assessment (HTA) as part of the acceptance and uptake of a new medicine into a healthcare system (HTA is covered specifically in Topic ‘HTA Principles and Practices’, Course 1). The overall goal usually is a global development programme with simultaneous, multimarket submissions and approvals.
There is general consensus that such development requires the collaborative effort of a multidisciplinary and cross-functional team of experts who will establish the global development framework, incorporating data and regulatory requirements and strategies to obtain a marketing authorisation in the shortest possible timeframe. It is evident that the regulatory input throughout is indispensable, not least because all evidence needed for a marketing authorisation application (or variation of a MA) will ultimately be processed and incorporated in an application managed by the regulatory affairs function. A high-level graphic representation of this approach is shown in Figure 2.
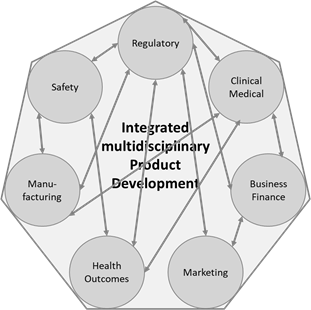
Figure 2: High level schematic of the integrated, multidisciplinary medicines development. Arrows indicate exemplary the main bidirectional information flow between the different actors during the process.
Therefore, it is a prerequisite for their work that regulatory professionals:
- hold a broad overview on the full set of medicines regulations pre- and post-marketing and equally of the entire development process
- understand the organisation’s business strategy to plan and execute the product development and post authorisation regulatory activities within this framework and as members of a multidisciplinary team.