Completion requirements
View
1. Legal bodies responsible for regulatory procedures
1.2. European Medicines Agency (EMA)
The European Medicines Agency (EMA), established in 1995:
- underpins the centralised authorisation procedure and supports coordination between NCAs. The Agency is the hub of a European medicines network comprising over 40 national regulatory authorities.
- is a decentralised agency of the European Union acting as a networking organisation based in Amsterdam, whose activities involve thousands of experts from across Europe. These experts carry out the work of EMA's scientific committees
- facilitates development and access to medicines
- performs the scientific evaluation of applications for marketing authorisation under the Centralised Procedure (CP).
- supervises and monitors the safety of medicines in the EU across their lifecycle
- provides information to healthcare professionals and patients and publishes clear and impartial information about medicines and their approved uses. This includes public versions of scientific assessment reports and summaries written in lay language.
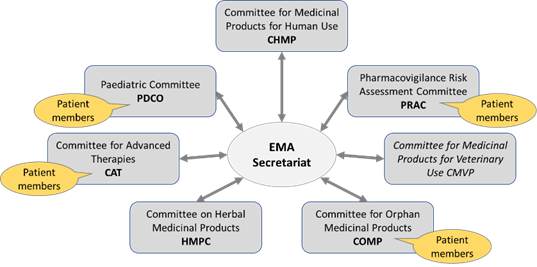
- has seven scientific committees*, six of which concerned with human medicines, that perform its scientific assessments and evaluate medicines along their lifecycle from early stages of development, through marketing authorisation to safety monitoring once they are on the market.
- has in addition a number of working parties and related groups which the committees can consult on scientific issues relating to their particular field of expertise
EMA’s scientific committees:
It may be of interest to also know what is NOT in the remit of EMA. The box below (adapted from the EMA website) contains those items which EMA lists as not falling under their activities.
What EMA does not do
- evaluate the initial marketing authorisation application of all medicines in the EU. For more information on medicine authorisation routes in the EU, see Authorisation of medicines;
- evaluate applications for the authorisation of clinical trials. The authorisation of clinical trials occurs at Member State level, although the Agency manages a database of clinical trials carried out in the EU.
- evaluate medical devices. Medical devices are regulated by national competent authorities in Europe. EMA is involved in the assessment of certain categories of medical devices. (e.g. an ingestible sensor that is incorporated into a medicinal product)
- evaluate food supplements and cosmetics. These substances are evaluated at national level;
- carry out research or develop medicines. Pharmaceutical companies or other medicines developers carry out the research and development of medicines, who then submit the findings and test results for their products to the Agency for evaluation;
- take decisions or have information on the price or availability of medicines. Decisions about price and reimbursement take place at the level of each Member State in the context of the national health system of that country;
- control the advertising of medicines. The control of the advertising of non-prescription medicines in the EU is primarily conducted on a self-regulatory basis by industry bodies, supported by the statutory role of the national regulatory authorities in the Member States;
- control or have information on pharmaceutical patents. Patents having effect in most European countries may be obtained either nationally, via national patent offices, or via a centralised process at the European Patent Office;
- develop treatment guidelines. National governments or the health authorities of individual EU Member States develop guidelines for decisions regarding diagnosis, management, and treatment in specific areas of healthcare (sometimes known as clinical guidelines);
- provide medical advice;
- develop laws concerning medicines. The European Commission develops EU legislation concerning medicines and the European Parliament together with the Council of the European Union adopt it. The European Commission also develops EU policies in the field of human or veterinary medicines and public health. For more information see European Commission: Medicinal products for human use;
- issue marketing authorisations. The legal decision to grant, suspend or revoke a marketing authorisation for any medicine falls under the remit of the European Commission for centrally authorised products, and the national competent authorities of the EU Member States for nationally authorised products