Completion requirements
View
4. Stakeholders in pharmacovigilance
Key stakeholders directly involved in pharmacovigilance activities include:
- Regulatory authorities, including
- the European Commission (EC) is the competent authority for centrally authorised products (CAP) and the legal authority that underpins the EU PhV system andprovides the legislative framework needed to carry out its functions in the most efficient way.
- the European Medicines Agency (EMA): central role in the EU system, co-ordinating its activities and providing technical, regulatory and scientific support to the Member States (MS) and industry.
- the National Competent Authorities (NCAs) (member states regulatory authorities): organise and supervise information collection on and reporting of suspected adverse reactions of medicines, particularly spontaneous reports from patients and health care professionals (HCP) (required by law[1] ); take the lead (rapporteur and co-rapporteur teams) in evaluating and analysing data when a safety issue is assessed at the European level (a PhV referral); etc.
- the Pharmacovigilance Risk Assessment Committee (PRAC): an EMA scientific committee in the revised European PhV system (since 2012); members include experts in PhV and regulation working within the NCAs, representatives of patients and HCPs, and scientific experts in areas such as epidemiology, signal detection, biological medicines and risk communication nominated by the EC; responsible for the assessment of safety issues at EU level; also monitors many of the PhV activities. It is at the core of the EU signal management process.
The medicines regulatory authorities in 31 European Economic Area (EEA)
countries, the EMA and the European Commission closely collaborate and work in
partnership as a network (European medicines regulatory network) to
discuss and deal swiftly with any emerging problem in the interest of patients'
access to safe and efficacious medicines.
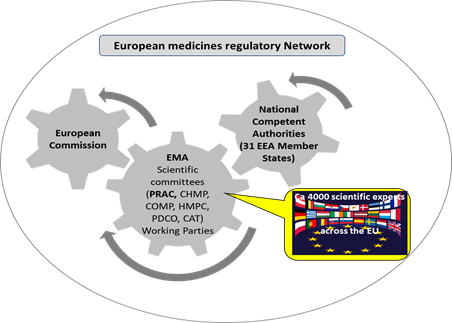
Figure 2: Simplified schematic of the European medicines regulatory network
- Patients and patient organisations (PO):
- POs work with some NCAs to facilitate adverse reaction reporting; number of POs involved per Member State varies from 1 to 20;
- user-test adverse reaction reporting forms;
- review relevant safety communications (see lesson 4);
- patient representatives are members of PRAC; have input to all activities of the Committee; supply relevant perspectives to all aspects of its work
- broader consultations may form part of PhV referrals
- patient representatives participate in
scientific advisory groups (SAGs), (expert groups convened to supply
specialist input)
- Health care professionals (HCP) (Doctors, pharmacists, nurses and all other professionals working with medicines):
- are members of PRAC; participate in all activities of the Committee; supply relevant perspectives to all aspects of its work;
- broader consultations may form part of PhV referrals;
- review relevant safety
communications
- the Pharmaceutical industry: individual marketing authorisation holders (MAHs) have specific responsibilities under the legislation in terms of running a system of PhV, monitoring and reporting for their products. MAHs are responsible for collecting, reviewing and analysing information on suspected adverse reactions received via patients, healthcare professionals, scientific literature, clinical trials and studies or other sources. They have to report this information to regulatory authorities for further evaluation on an expedited basis (via sharing suspected adverse reaction reports as well as any urgent safety issues identified) and on a periodic basis (via Periodic Safety Update Reports (PSURs)). MAHs communicate and collaborate with regulators as appropriate in dealing with PhV issues.