4. EudraVigilance
| Stránky: | EUPATI Open Classroom |
| Kurz: | Pharmacovigilance - Risk management |
| Kniha: | 4. EudraVigilance |
| Vytiskl(a): | Nepřihlášený host |
| Datum: | čtvrtek, 23. července 2026, 09.07 |
Obsah
- 1. EudraVigilance
- 1.1. EudraVigilance system: users and data sources
- 1.2. Reports of suspected ADRs and other information
- 1.3. Reports
- 1.4. The EudraVigilance database containing “Article 57 database”
- 1.5. EudraVigilance webportal: adrreports.eu
- 1.6. Public Access to EudraVigilance
- 1.7. Access to data held in EudraVigilance
- 1.8. Public Access to EudraVigilance
1. EudraVigilance
(This section is organised in the form of a book, please follow the blue arrows to navigate through the book or by following the navigation panel on the right side of the page.)
EudraVigilance (EV) is a centralised European Union (EU) system for managing and analysing information on suspected adverse reactions (ADR) to a medicine. It is the tool that the EMA and NCAs use for the monitoring of the safety of all medicines authorised or being studied in clinical trials in the European Economic Area (EEA). It was created and is operated by the European Medicines Agency and has been in use since December 2001.
The EudraVigilance system includes a fully automated safety and message-processing mechanism and a large centralised pharmacovigilance database with query and tracking functions, which serves the early detection of potential safety signals and their evaluation. EudraVigilance also incorporates the Medical Dictionary for Regulatory Activities (MedDRA).
- EudraVigilance is composed of the following main system components supporting functions:
- Data processing and management system components
- EudraVigilance Gateway, a data-processing network for the secure electronic exchange of ADR data.
- EudraVigilance Post-Authorisation Module (EVPM) for collection of Individual Case Safety Reports (ICSRs) (a document providing information related to an individual case of a suspected adverse reaction due to a medicine) related to all medicinal products authorised in the EEA (Regulation (EC) No 726/2004 and Directive 2001/83/EC). The following ICSR types are collected in EVPM: “Spontaneous Report”, “Report from Study” with study type: “Individual patient use” (e.g., ‘compassionate use’ or ‘named patient basis’) and “Other studies”, “Other” (e.g., pharmacoepidemiology, pharmacoeconomics, intensive monitoring) and “Not available to sender (unknown)”;
- EudraVigilance Clinical Trial Module (EVCTM) for collection of ICSRs of Suspected Unexpected Serious Adverse Reactions (SUSARs) (Directive 2001/20/EC and Regulation (EU) No 536/2014 on clinical trials). The following ICSR types are collected in EVCTM: “Report from Study” with study type: “Clinical Trials” (interventional studies).
- eXtended EudraVigilance Medicinal Product Dictionary (XEVMPD), reference source for coding
Note: EVPM and EVCTM can be accessed by registered users through EVWEB, a web interface with a set of functionalities to aid the creation, electronic reporting of and access to ICSRs. EVWEB includes the ICSR Export Manager, which permits the download of ICSRs in the internationally agreed format.
Data analysis and signal detection component
- EudraVigilance Data Warehouse (pharmacovigilance database) and Analysis System (EVDAS), to support the EU pharmacovigilance safety monitoring activities with the main focus on signal detection and evaluation of ICSRs.
Adrreports.eu portal
- The portal allows to search and view data on suspected adverse reactions for authorised medicinal products in the EEA and provides general information to aid the understanding of the reports.
1.1. EudraVigilance system: users and data sources
Figure 1 gives a high-level system overview of EudraVigilance. Further details are provided in the following sections.
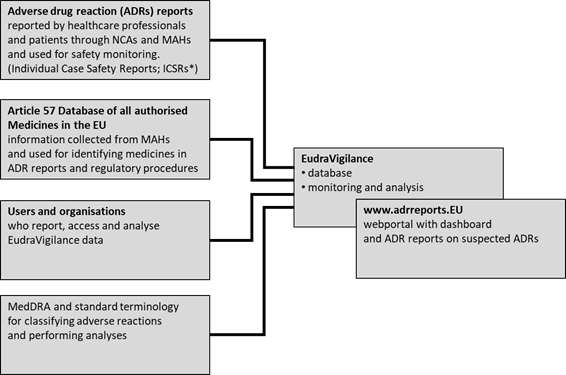
Figure 1. EudraVigilance users, data sources and data use.
(adapted from EMA
2019-annual-report-eudravigilance)
*[ICSR: a document providing information related to an
individual case of a suspected adverse reaction due to a medicine]
1.2. Reports of suspected ADRs and other information
The reporting of suspected ADRs in a consistent way across the European Economic Area (EEA) is very important. Timely detection and assessment of safety signals from sources such as EudraVigilance complements the benefit-risk-balance evaluation of periodic safety update reviews and the assessment of risk management plans (RMPs) by the PRAC. EudraVigilance is therefore one of the cornerstones of EU pharmacovigilance as it helps to:
- monitor the benefits and risks of medicines,
- detect emerging safety signals
- and decide on and initiate appropriate action.
|
In short, a safety signal can be defined as new information related to a known ADR or any other event, either adverse or beneficial, and potentially causally related to the use of a medicine, that requires further investigation. See also lesson 1 for a detailed definition. |
By facilitating electronic exchange of ICSRs between
EMA, NCAs, MAHs and sponsors of clinical trials in the EEA, EudraVigilance is
instrumental in ensuring the safety of medicines and early detection and
evaluation of possible safety signals.
1.3. Reports
1.3.1 Electronic reporting
Data in EudraVigilance is submitted electronically by national regulatory authorities and by marketing authorisation holders for medicines. Electronic reporting and evaluation of suspected ADRs is obligatory for MAHs and NCAs[1] and covers elements as shown in the following table (Table 1):
Table 1: Electronic reporting obligations in the EU; adapted from Eudravigilance-electronic-reporting
|
From where |
Who |
What |
|
Interventional clinical trials |
· Marketing authorisation holders
|
Reports of suspected unexpected serious adverse reactions (SUSARs) via safety reports in clinical trials (individual case safety reports (ICSRs)) |
|
• Marketing authorisation holders • Sponsors of clinical trials |
Information on investigational medicinal products via product reports (for Article 57 database) |
|
|
Spontaneous reports [2] and non-interventional studies |
|
Reports of suspected adverse reactions for authorised medicinal products via safety reports (ICSRs): - suspected serious adverse reactions occurring within and outside the EEA - suspected non-serious adverse reactions occurring within the EEA |
|
Marketing authorisation holders |
Information on authorised medicinal products via product reports (for Article 57 database) |
Patients, consumers and healthcare professionals (HCPs) can report, and are encouraged to do so, suspected adverse reactions to either the national competent authorities or the marketing authorisation holders of the medicine. These reports are then transmitted electronically and stored in the EudraVigilance database.
Background
Recognition by health authorities of the advantages of complementing reports on suspected adverse reactions from HCPs with those from patients triggered a number of changes to existing pharmacovigilance systems in Europe. In 2003, Denmark and the Netherlands became the first countries to allow patients and consumers to report suspected adverse reactions directly to their regulatory authority, followed by Italy (2004), the United Kingdom (2005) and Sweden (2008)[3] An example from the UK is shown in the following figure (Fig. 2)

Since 2012, with new EU pharmacovigilance legislation, patient reporting has been expanded throughout the EU. Member States are now mandated to encourage patients to report suspected adverse reactions directly to the regulatory authority and to enable reporting through web-based formats and alternative means. Likewise, marketing authorisation holders (MAHs) shall record all suspected adverse reactions, whether reported spontaneously by patients or healthcare professionals and not refuse to consider adverse reaction reports received from patients through appropriate means and shall ensure that those reports are accessible at a single point within the Union.[4]
By introducing a legal right for patients to report suspected ADRs directly to regulatory authorities, the EU acknowledges patients and consumers as key sources of information on medicines safety and fosters faster and more comprehensive collection of data on adverse reactions.
To note:
In spite of the fact that spontaneous reporting is crucial for signal detection, under-reporting is common. It is estimated that only 1% to 10% of serious adverse reactions are reported [5] . Healthcare professionals often attribute low reporting to lack of time and sometimes only report adverse reactions when convinced of a causal association between a medicine’s use and an adverse reaction [6]. Additionally, whilst patients consider certain ADRs to be very significant in affecting their quality of life, healthcare professionals do not to the same extent [7] .
The added value of patients’ reporting is that their reports tended to be more elaborate in description of suspected ADRs by including detailed descriptions of symptoms and the social, emotional and occupational impact. According to an evaluation of patient reporting to the Yellow Card Scheme [8], patients report a different spectrum of reaction types compared with healthcare professionals, although there is a reasonable amount of overlap. Because the patient focus in the reports differs from healthcare professionals, patient reporting of suspected ADRs has the potential to add considerable value to pharmacovigilance. They can generate new potential safety signals and describe suspected ADRs in enough detail to provide useful information on the likely causality and the impact on patients' lives.
Patients are more likely than healthcare professionals to report:
- Symptoms (93% vs 78%).
- Impact of the ADR (47% vs 12%).
- Temporal relationship between medicine and suspected ADR.
- Extreme nature of the suspected ADR (47% vs 17%).
Pharmaceutical companies holding or applying for a marketing authorisation in the European Economic Area (EEA), sponsors of clinical trials and NCAs in the EEA need to register with EudraVigilance for the electronic data interchange of pharmacovigilance information (see box below). The registration process is a prerequisite also for electronic reporting. All users, including organisations, need an active EMA account created through the EMA Account Management portal.
|
Registration is mandatory for:
|
[1]
DIRECTIVE 2001/83/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 6 November 2001 on the Community code relating to medicinal products for human use (consolidated 2019) Art.107, 107a.
https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02001L0083-20190726&from=EN
[2] Spontaneous reports are unsolicited reports by healthcare professionals or patients that do not derive from a study or any organised data-collection scheme. https://www.adrreports.eu/en/background.html
[3] Herxheimer, A, R Crombag, and TL Alves. "Direct f reporting of adverse drug reactions. A twelve-country survey; literature review." Health Action International Europe. May 2010.https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02001L0083-20190726&from=EN
[6] Herdeiro, Maria T et al. "Physicians’ Attitudes to Adverse Drug Reaction Reporting. A case control study in Portugal" Drug safety 28.9 (2005): 825-833.
[7] Frankenfeld, Christian. "“Serious” and “severe” adverse drug reactions need defining." BMJ 329.7465 (2004): 573.
[8] Avery AJ, Anderson C, Bond CM, Fortnum H, Gifford A, Hannaford PC, et al. Evaluation of patient reporting of adverse drug reactions to the UK Yellow Card Scheme: literature review, descriptive and qualitative analyses, and questionnaire surveys. Health Technol Assess 2011; 15 (20).http://www.ncbi.nlm.nih.gov/pubmed/21545758
1.4. The EudraVigilance database containing “Article 57 database”
The submission of data on medicines by MAHs is a legal requirement from the 2010 pharmacovigilance legislation. Data in EudraVigilance is submitted electronically by NCAs and by pharmaceutical companies that hold the marketing authorisation for medicines. The aim is to establish a complete inventory of all medicines authorised for use in the EU and EEA, including both, centrally authorised medicines via the EMA and those authorised at national level. MAHs are required to submit information on their medicines to the Article 57 database in accordance with Article 57(2) of Regulation (EC) No. 726/2004.The EMA uses this information for:
- analysis of data in EudraVigilance and signal management;
- reporting and coding of individual case safety reports;
- facilitating medicines regulation and fulfilling regulatory actions and legal obligations;
- and strengthening communication with stakeholders.
EMA publishes information on all authorised medicines contained in the Article 57 database in the form of an excel document.
The document contains the following data fields:
- product name (product short name: brand name or the combination of the generic name and the company name);
- active substance;
- route of administration;
- country of authorisation;
- name of the marketing authorisation holder (company);
- country of location of the pharmacovigilance system master file;
- marketing authorisation holder's contact email address and telephone number for pharmacovigilance enquiries.
Only
products that have a valid marketing
authorisation are included in the document and
EMA updates this document periodically to reflect changes in the Article 57
database.
Below the link to an example for this information:
Article 57 product data (XLSX/7.31 MB) (updated)
First published: 30/07/2018
Last updated: 02/06/2021
EMA/518502/2018 Rev. 35
The EudraVigilance database holds over 16 million Individual Case Safety Reports (ICSRs) referring to over 9 million cases and is one of the largest pharmacovigilance databases in the world. Enhanced functionalities, following significant recent development, allow for a better support of pharmacovigilance activities and the protection of public health. In other jurisdictions similar approaches exist.
1.5. EudraVigilance webportal: adrreports.eu
EudraVigilance data are published via the webportal adrreports.eu in 26 languages and provide information on reports of suspected adverse reactions (Individual Case Safety Reports, (ICSRs)) for authorised medicines in the European Economic Area (EEA). The output formats include web reports containing aggregated information, using data elements from the reports submitted to EudraVigilance, line listings providing an overview in tabular format about individual cases, whilst individual report forms present more detailed information specific to a case.
This web portal was launched by the EMA in 2012 to comply with the EudraVigilance access policy, developed to improve public health by supporting the monitoring of the safety of medicines and to increase transparency for stakeholders, including the general public. The policy is designed to provide as much information as possible while meeting data protection obligations. A revision of the access policy “Revision 4: update of references in accordance with Regulation (EU) 2016/679, the General Data Protection Regulation (GDPR) and Regulation (EU) 2018/1725, the EU Data Protection legislation (EU DPR)” took place in 2019.
Subsets of data (defined sets of ICSR data elements) from spontaneous reports [1] listed in Annex B of the EudraVigilance Access Policy, are released, taking into account the EU DPR. This applies to all types of medicines, independent of the authorisation procedure and the source of the report (e.g., healthcare professional, patient).
For centrally authorised medicines, reports can be accessed both by the name of the medicine or the name of the active substance or combination thereof, for non-centrally authorised medicines, by the name of the active substance.
To note:
The information from this web portal relates to suspected adverse
reactions, i.e. effects that have been observed following administration of, or
treatment with, a medicine. However, these
suspected adverse reactions may not be related to, or caused by the medicine
or active substance.
Therefore information in a web report cannot be used to determine the likelihood of experiencing a particular adverse reaction caused by the medicine in question. The information cannot be used in isolation to decide upon the benefit-risk balance of a medicine. Other information, such as how many people take the medicine and how long it has been on the market, needs to be considered. Moreover, apart from the spontaneous reports arising from the use of a medicine worldwide, information can also be gathered from additional sources, such as clinical trials and epidemiologic studies, scientific literature, rates of morbidity (incidence of disease in a population) and of mortality (incidence of death in a population). Sometimes, additional investigative studies, and consultations with other medicines regulatory authorities, are necessary to assess the likelihood that a suspected adverse reaction is linked to a medicine, to identify risk factors, and to estimate the frequency of occurrence. Only the assessment of all available data allows for robust conclusions on the benefits and risks of a medicine to be drawn. In essence, any individual case report should be seen in the context of all available data on the medicine.
[1] Spontaneous reports are unsolicited reports by healthcare professionals or patients that do not derive from a study or any organised data-collection scheme.
1.6. Public Access to EudraVigilance
The adrreport.eu web portal is the gateway to access information from EudraVigilance. This is of specific interest for stakeholder group II, i.e., healthcare professionals and the public.
Since as already mentioned not all data elements can be made available publicly, the following table gives an overview of data elements released for stakeholder group II and from which section of the database.
Table 3: Overview of accessible data elements for stakeholder group II (HCPs, public) and respective EudraVigilance sections and explanatory notes (adapted from EudraVigilance access policy and EudraVigilance User Manual for online access (Version 2.0))
| Stakeholder Group II Healthcare Professionals and the Public | ||
|
Access point:
|
Content overview
|
|
| Section according to ICH E2B(R3) ICSR Implementation Guide | Data element name | Explanation/possible values |
| C.1 Identification of the Case Safety Report | Type of Report “EudraVigilance Local Report Number” |
classification of a report by sender (e.g., spontaneous report) displayed as “EudraVigilance Local Report Number” |
| C.2.r Primary Source(s) of Information | Reporter’s Country Code Qualification (reporter group) |
Country code displayed as EEA or Non-EEA Healthcare Professional (Physician, Pharmacist or Other HCP) Non-Healthcare Professional (Lawyer, Consumer or Other non-HCP) |
| C.3 Information on Sender of Case Safety Report | Sender Type Sender’s organisation |
Displayed as e.g. "EEA Regulator" or “Regional Pharmacovigilance Centre” |
| C.4.r Literature Reference(s) | Literature Reference(s) | |
| C.5 Study Identification | Study Registration Number Study Name Study Type Where Reaction(s) / Event(s) were Observed |
|
| D. Patient Characteristics | Patient Age Group (as per reporter) Sex |
Age group (mapped against “Age at Time of Onset of Reaction/Event”, based on the reported patient age or calculated based on difference between “Date of Birth” and “First Reaction Start Date”) Male/female/not specified |
| E.i Reaction(s)/event(s) | Reaction / Event (MedDRA codeE) | |
| Results in Death Life Threatening Caused / Prolonged Hospitalisation Disabling / Incapacitating Congenital Anomaly / Birth Defect |
Serious | |
| Other Medically Important Condition (Non-Serious) Suspected Reported Reaction (Preferred Term in MedDRA) (PT) Reaction Group (System Organ Class in MedDRA) (SOC) |
Any undesirable effect (suspected adverse reaction) reported by the reporter e.g. Headache, Ear infection e.g. Nervous system disorders |
|
| Duration of reaction / Event (number) Duration of Reaction / Event (unit) |
||
| Outcome of Reaction / Event at the Time of Last Observation | Recovered/resolved Recovering/resolving Not recovered/not resolved Recovered/resolved with sequelae Fatal Unknown Not specified |
|
| G.k Drug(s) information | Characterisation of Drug Role Pharmaceutical Product Identifier (PhPID) Medicinal Product Name as Reported by the Primary SourceF Substance / Specified Substance NameF |
|
| G.k.4.r Dosage and Relevant Information | Dose (number) Dose (unit) Number of Units in the Interval Definition of the Time Interval Unit Duration of Drug Administration (number) Duration of Drug Administration (unit) Pharmaceutical Dose Form Term ID Route of Administration Term ID Parent Route of Administration Term ID |
|
| G.k.7.r Indication for Use in Case | Indication (MedDRA code) Action(s) Taken with Drug |
|
| G.k.9.i Drug-reaction(s) / Event(s) Matrix | Did Reaction Recur on Re-administration? Additional Information on Drug (coded) |
|
Users can search by defined parameters (e.g. product (medicines), active substance, reaction, age group, gender, time period)
There are ‘Download’ and ‘Print’ functions for search results. This might be a summary or individual reports based on defined data elements ensuring compliance with personal data protection legislation. Users are provided with clear guidance on interpretation of the data.
Of the presently 272 data elements per report, for stakeholder group II access will be granted to 53 of the data elements.
It also needs to be recognised that not all data elements of ICSRs are always completed. This means that although access is provided to certain data elements, information may not always be available given the type of the report or the primary source of the information.
Public access to aggregated EudraVigilance data: In November 2017 public access via www.adrreports.eu was further improved by providing additional outputs such as line listings and individual case report forms. By the end of 2019, the website provided information on a total of 3,724 active substances, of which 753 contained in centrally authorised products (CAPs) and 2,971 in nationally authorised products (NAPs).
1.7. Access to data held in EudraVigilance
Different stakeholders such as NCAs in EEA countries, healthcare professionals, patients and consumers, as well as MAHs and academia can access data held in the EudraVigilance database. Access is defined based on the stakeholder's interests and needs as well as the requirement to comply with applicable personal data protection legislation.
Therefore, the access is subdivided in different levels, taking into consideration that, due to the often-detailed nature of the information, not all data elements can be disclosed to avoid a potential re-identification of data subjects. Six stakeholder groups are defined, following the EudraVigilance access policy rules. (see 4.4 (2))
The table below gives an overview of the different stakeholder groups, their data access and access provisions.
Table 2: Overview of stakeholder groups and their access to data elements in EudraVigilance. (adapted from EudraVigilance access policy and EudraVigilance User Manual for online access (Version 2.0))
|
Stakeholder Group (SG) |
EudraVigilance(EV) Data Access |
Access Pre-requisites & Mechanisms |
|
SG I Medicines regulatory authorities in the EEA, the EMA and EC |
|
EC and EMA: Registration with password -protected EudraVigilance access. NCAs: via EVDAS, incl. data analysis and signal detection; |
|
|
|
|
|
SG II Healthcare Professionals and the Public |
All spontaneous reports as aggregated data and line listings and ICSR forms based on restricted data elements. |
adrreports.eu web portal; no authorisation for accessing the ICSR data set is required. |
|
|
|
|
|
SG III Marketing-authorisation holders |
|
Registration with password-protected access to EV
Confidentiality undertaking for access to case narratives for signal validation or other PV assessment procedure |
|
|
|
|
|
SG IV Academia |
|
adrreports.eu web portal
Confidentiality undertaking and provision of ad-hoc data set prepared by the EMA |
|
|
|
|
|
SG V WHO Uppsala Monitoring Centre |
|
Data transfer agreement and daily electronic data transfer |
|
|
|
|
|
SG VI Medicines regulatory authorities outside the EU |
|
adrreports.eu web portal Ad-hoc data set prepared by the EMA |
1.8. Public Access to EudraVigilance
The adrreport.eu web portal is the gateway to access information from EudraVigilance. This is of specific interest for stakeholder group II, i.e., healthcare professionals and the public.
Since as already mentioned not all data elements can be made available publicly, the following table gives an overview of data elements released for stakeholder group II and from which section of the database.
Table 3: Overview of accessible data elements for stakeholder group II (HCPs, public) and respective EudraVigilance sections and explanatory notes (adapted from EudraVigilance access policy and EudraVigilance User Manual for online access (Version 2.0))
|
Stakeholder Group II Healthcare Professionals and the Public |
||
|
Access point: · Adrreports.eu portal |
Content overview · Aggregated data outputs based on predefined queries · ICSR line listings (based on core ICSR data elements) · ICSR forms (for individual case review) |
|
|
Section according to |
Data element name |
Explanation/ |
|
C.1 Identification of the Case Safety Report |
Type of Report “EudraVigilance Local Report Number” |
classification of a report by sender (e.g., spontaneous report) displayed as “EudraVigilance Local Report Number” |
|
C.2.r Primary Source(s) of Information |
Reporter’s Country Code Qualification (reporter group) |
Country code displayed as EEA or Non-EEA Healthcare Professional (Physician, Pharmacist or Other HCP) Non-Healthcare Professional (Lawyer, Consumer or Other non-HCP) |
|
C.3 Information on Sender of Case Safety Report |
Sender Type Sender’s organisation |
Displayed as e.g. "EEA Regulator" or “Regional Pharmacovigilance Centre” |
|
C.4.r Literature Reference(s) |
Literature Reference(s) |
|
|
C.5 Study Identification |
Study Registration Number Study Name Study Type Where Reaction(s) / Event(s) were Observed |
|
|
D. Patient Characteristics |
Patient Age Group (as per reporter) Sex |
Age group (mapped against “Age at Time of Onset of Reaction/Event”, based on the reported patient age or calculated based on difference between “Date of Birth” and “First Reaction Start Date” Male/female/not specified |
|
E.i Reaction(s)/event(s) |
Reaction / Event (MedDRA codeE) |
|
|
Results in Death Life Threatening Caused / Prolonged Hospitalisation Disabling / Incapacitating Congenital Anomaly / Birth Defect |
Serious
|
|
|
Other Medically Important Condition (Non-Serious) Suspected Reported Reaction (Preferred Term in MedDRA) (PT) Reaction Group (System Organ Class in MedDRA) (SOC) |
Any undesirable effect (suspected adverse reaction) reported by the reporter e.g. Headache, Ear infection
|
|
|
Duration of reaction / Event (number) Duration of Reaction / Event (unit) |
|
|
|
Outcome of Reaction / Event at the Time of Last Observation |
Recovered/resolved Recovering/resolving Not recovered/not resolved Recovered/resolved with sequelae Fatal Unknown Not specified |
|
|
G.k Drug(s) information |
Characterisation of Drug Role Pharmaceutical Product Identifier (PhPID) Medicinal Product Name as Reported by the Primary SourceF Substance / Specified Substance NameF |
|
|
G.k.4.r Dosage and Relevant Information |
Dose (number Dose (unit)) Number of Units in the Interval Definition of the Time Interval Unit Duration of Drug Administration (number) Duration of Drug Administration unit) Pharmaceutical Dose Form Term ID Route of Administration Term ID Parent Route of Administration Term ID |
|
|
G.k.7.r Indication for Use in Case |
Indication (MedDRA code) Action(s) Taken with Drug |
|
|
G.k.9.i Drug-reaction(s) / Event(s) Matrix |
Did Reaction Recur on Re- administration? Additional Information on Drug (coded |
|
Users can search by defined parameters (e.g. product (medicines), active substance, reaction, age group, gender, time period)
There are ‘Download’ and ‘Print’ functions for search results. This might be a summary or individual reports based on defined data elements ensuring compliance with personal data protection legislation. Users are provided with clear guidance on interpretation of the data.
Of the presently 272 data elements per report, for stakeholder group II access will be granted to 53 of the data elements.
It also needs to be recognised that not all data elements of ICSRs are always completed. This means that although access is provided to certain data elements, information may not always be available given the type of the report or the primary source of the information.
Public access to aggregated EudraVigilance data: In November 2017 public access via www.adrreports.eu was further improved by providing additional outputs such as line listings and individual case report forms. By the end of 2019, the website provided information on a total of 3,724 active substances, of which 753 contained in centrally authorised products (CAPs) and 2,971 in nationally authorised products (NAPs).