2. About Good 'x' Practice (GxP)
| Site: | EUPATI Open Classroom |
| Course: | Introduction to Regulatory Affairs |
| Book: | 2. About Good 'x' Practice (GxP) |
| Printed by: | Guest user |
| Date: | Friday, 27 June 2025, 6:53 PM |
1. About Good 'x' Practice (GxP)
(This section is organised in the form of a book, please follow the blue arrows to navigate through the book or by following the navigation panel on the right side of the page.)
Regulatory frameworks apply throughout the entire product lifecycle, from early-stage development to post authorisation. GxP is an acronym that refers to the ‘good practice’ laws, rules and guidelines applicable to organisations that develop, test, manufacture and market or distribute products that are consumed or used by humans such as medicinal products [1] (hereinafter medicines), medical devices, medical software applications, cosmetics, or food products.
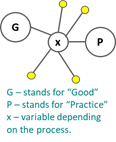
The “x” is the variable in GxP and covers a wide range of processes in the development, manufacture, and distribution of products. This lesson concentrates on the pharmaceutical area and deals with GxPs related to medicines predominantly.
The underlying quality rules and guidelines in the respective GxPs define the various ways companies in this highly regulated industry are required to ensure that their products for human use are of suitable quality and are safe, effective and usable . When companies deploy computerised systems for certain GxP processes, they must also implement a computerised GxP system, validated, and operated appropriately for the intended use of the system.
The main concepts that all GxP regulations share:
- Traceability: the ability to reconstruct a product’s entire development (or lifecycle) history, as well as that of each and every work item that contributed to its development.
- Accountability: knowing who was involved in the product’s development and manufacture, including when and how exactly did they contribute.
- Data integrity: a Quality Management system, called “Pharmaceutical Quality System (QPS)”, ensuring Attributable, Legible, Contemporaneous, Original, Accurate (ALCOA), complete, consistent, enduring, and available (ALCOA+ [1] ) data.
Of note: an increasingly used term for GxP is cGxP which stands for “currentGxP”. It means the current good manufacturing, distribution and other good practices specified by the US Code of Federal Regulations, the EU, PIC, ICH and WHO, and, as applicable, by any other Regulatory Authority. It represents the current thinking of the respective regulatory authority on a topic
[1] ALCOA is an acronym that defines the fundamentals for ensuring data integrity and critical elements of Good Documentation Practice (See section 2.6. ALCOA stands for “attributable, legible, contemporaneous, original and accurate”. ALCOA-plus (ALCOA+) adds additional emphasis on the attributes of being “complete, consistent, enduring and available”. Used by WHO, EMA, FDA, PIC/S(Pharmaceutical Inspection Co-Operation Scheme) and others
[1] The term "Medicinal Product" in the European Union approximately corresponds to the term "Drug Product" in the United States. Sometimes the term "Finished Product" is used instead.
The term "Active Substance" or “Active Pharmaceutical Ingredient (API) in the European Union corresponds to "Drug Substance" in the United States. Within EUPATI the term “drug” is used for “substance” or API.
1.1. GxP Regulatory oversight
Different countries/regions have their own defined GxP rules and guidelines, enforced and audited by the respective regulatory authority, and particular GxP criteria covering specific topics can be found in legislation and guidelines and also industry best-practice frameworks. Nevertheless, the fundamental concept and requirements of GxP are similar across countries/regions; they have almost uniformly been adopted by government agencies, e.g., related to product manufacturing processes, documentation procedures, staff qualifications and training, distribution, etc.
In the EU, GxP regulations are defined in the European Commission[1] EudraLex[2]. In the US, GxP regulations in the Food & Drug Administration (FDA) Code of Federal Regulations (CFR) Title 21[3] or in Japan; in the Japan’s Ministry of Health, Labour and Welfare (MHLW) “Act on Securing Quality, Efficacy and Safety of Pharmaceuticals, Medical Devices, Regenerative and Cellular Therapy Products, Gene Therapy Products, and Cosmetics” (abbreviated the PMD Act[4]). In many countries, the PIC/S (Pharmaceutical Inspection Co-operation Scheme) GMP[5] and the ICH (The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use) guidelines[6] are adapted.
The respective regulatory authorities are: the European Medicines Agency (EMA) for Europe, the FDA for the USA, and Japan’s Pharmaceuticals and Medical Devices Agency (PMDA).
It is nevertheless important to note that national guidelines can have a major impact on companies beyond borders. Regulators running GxP inspections in other jurisdictions expect that their own standards are followed. The proliferation of mutual recognition agreements, such as that between the U.S. and the EU, means that what appear to be local GMP issues can quickly become international. Local breaches of Good Laboratory Practice (GLP) and Good Clinical Practice (GCP) can mean that the results of non-clinical and clinical studies become ineligible for submission to regulators in any jurisdiction. Therefore, companies need to comply with differing standards of Good Pharmacovigilance Practice (GVP) from another jurisdiction.
[3] FDA GMP: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm
FDA GDP, GLP:https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm
[4] Japan Act on Securing Quality, Efficacy and Safety of Products Including Pharmaceuticals and Medical Devices: https://www.pmda.go.jp/files/000152211.pdf
[5] PIC/S Guide GMP 2021: https://picscheme.org/docview/4205
[6] ICH Quality Guidelines: https://www.ich.org/page/quality-guidelines
1.2. Compliance and inspections
All organisations involved in the development, marketing, manufacture and distribution of medicines are responsible for ensuring that they comply with all relevant standards set out in European Union (EU) legislation and guidelines on pharmaceuticals. The GxP requirements outlined by the respective authorities, ask companies to formally define controlled processes tools and quality systems, irrespective of the operational sector and the specific GxP standards that apply, to demonstrate for example:
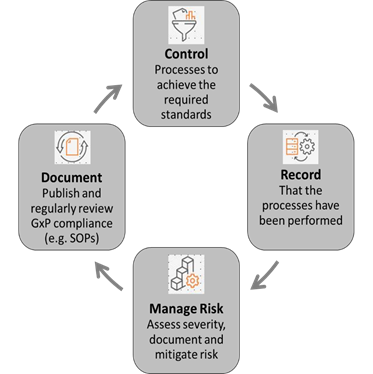
- How new health technologies are researched and developed
- How products are consistently manufactured
- How products are tested
- How laboratory and manufacturing equipment is calibrated and maintained
- How possible risks are identified and mitigated
- How processes are traced from start to finish through controlled documentation
- How different kinds of products are stored and distributed
- How those involved in the processes are trained
- How records of all these processes are captured and maintained to monitor continued effectiveness of all respective processes and systems
GxP compliance is monitored and enforced by agencies and government bodies through regular inspections and unannounced auditing, and certification requirements where applicable (Medical Devices). For companies this includes also to verify and vouch for the GxP compliance of their partners throughout the supply chain.
In many cases the scope of the regulator’s remit continues to widen taking in new sectors and product categories. This reflects the extent of innovation in these areas, as well as cross-sector dependencies in the techniques they use.
The European Medicines Agency (EMA) is responsible for harmonising the standards at EU level. The standards are constantly updated, developed and agreed, under the coordination of the EMA, by representatives of the GMP Inspectorates of each Member State. Guidelines on inspections of pharmacovigilance of medicinal products are part of the Good Vigilance Practice guidelines adopted by the EMA. Guidelines on GCP inspections are part of EudraLex, Volume 10.
The EMA coordinates inspections for medicines authorised under the centralised procedure or in the context of a referral, on request from the Committee for Medicinal Products for Human Use (CHMP).
EMA does not conduct inspections itself but inspections are carried out by the national competent authorities in the EU Member States. Inspections are performed regularly on sites within and outside the EU involved in developing, manufacturing and distributing medicines intended for the EU market, to verify their compliance with the relevant standards. An inspection may either be 'for cause', when it is triggered by a finding of possible non-compliance with relevant standards, or routine, when inspections are carried out as part of a surveillance programme.
Inspections are conducted both for authorised medicines and for medicines under evaluation in the EU. These inspections ensure that the rights, safety and wellbeing of clinical-trial participants are protected, and the reliability and integrity of data that support the authorisation of medicines and their quality, safety and effectiveness once on the market is duly controlled.
Legal basis:
Regulation
(EC) No 726/2004 Articles 8, 19, 57.1(i)
Council Directive 2001/83/EC Article 111
Member States are obliged to take account of the Compilation* by virtue of
Article 3(1) of Directive (EU) 2017/1572 and Article 17.1 of Regulation (EU)
2017/1569.
*Compilation of Union Procedures on Inspections and Exchange of Information
Table 1: Brief overview of inspections adapted from EMA https://www.ema.europa.eu/en/human-regulatory/overview/compliance-overview#inspections:-verifying-compliance-section
|
Inspections |
GCP |
GMP |
GLP |
GVP |
|
Purpose of inspections |
Confirms that clinical trials supporting a marketing authorisation application are conducted in line with GCP standards and all relevant EU legal and regulatory requirements |
Confirms that any medicine intended for the EU market is manufactured in compliance with GMP standards and all relevant EU legal and regulatory requirements |
Confirms that non-clinical studies supporting a marketing authorisation application are conducted compliant with GLP standards and all relevant EU legal and regulatory requirements |
Confirms that the marketing authorisation holder has personnel, systems and facilities to meet their pharmacovigilance obligations |
|
Sites in scope |
Any site where clinical trials that are part of an application are carried out, no matter where in the world it is located. |
· Any site manufacturing a medicine intended for the EU market, no matter where it is located · Sites in the EU are routinely inspected by the national authority · Typically performed at manufacturing sites located in third countries, for which a mutual recognition agreement (MRA) with the EU does not apply |
Any site where pivotal non-clinical studies are carried out, no matter where in the world it is located |
Any party carrying out pharmacovigilance activities in whole or in part, on behalf of or in conjunction with, the marketing authorisation holder, no matter where in the world it is located |
|
When they occur (on CHMP request) |
At any stage in the evaluation of a medicine. |
At any time during a medicine's evaluation or after authorisation in the EU. |
At any stage in the evaluation of a marketing authorisation application. |
At any stage during the evaluation of a marketing authorisation application or after a medicine is authorised. |
|
Impact of adverse outcomes |
If a GCP inspection reports critical or major findings on the conduct of clinical trials: · the CHMP evaluates the impact of the findings on the medicine’s benefit-risk balance and on the rights, safety and wellbeing of clinical trial participants · the CHMP may request analyses of clinical trial data excluding the affected patients or sites |
If a GMP inspection leads to adverse findings, the company must implement a corrective action plan agreed with the inspectors. |
If inspectors report critical or major findings on the conduct of non-clinical studies, the CHMP evaluates the impact on the medicine’s benefit-risk balance. |
|
|
Publicly available information |
Each medicine's assessment report includes information on the: · types and locations of sites inspected; · any impact of inspection findings on the benefit-risk assessment of the medicine. |
The EudraGMDP database holds the data collected during GMP inspections by EU authorities. Each medicine's assessment report includes information on the: · types and locations of sites inspected; · any impact of inspection findings on the benefit-risk assessment of the medicine. |
Each medicine's assessment report includes information on the: · types and locations of sites inspected; · any impact of inspection findings on the benefit-risk assessment of the medicine. |
|
|
International cooperation |
If the clinical trials took place outside the EU, and if EMA has a confidentiality arrangement with the responsible non-EU regulator, the two jurisdictions may: · exchange information on inspection findings and their interpretation; · conduct collaborative inspections. |
If a medicine is manufactured outside the EU, and if the EU has a mutual recognition agreement (MRA) with the country in question, the two jurisdictions can: · rely on each other's GMP inspection system; · share information on GMP inspections and quality defects;waive batch testing of products on import into their territories; EMA also takes part in international initiatives to: · exchange information on inspection planning and outcomes; · conduct joint inspections for manufacturing sites of common interest. |
Fpr non-clinical studies outside the EU, and if EMA has a confidentiality arrangement with the responsible non-EU regulator, the two jurisdictions may: · exchange information on inspection findings and interpretation; · conduct collaborative inspections. The EU is a signatory to the OECD mutual acceptance of data (MAD) agreement, which allows participating authorities to rely on each other's monitoring programmes for GLP compliance. |
EMA can exchange information on pharmacovigilance inspection findings with non-EU regulators with which EMA has signed a confidentiality arrangement. |