Early Clinical Development Plan
| Site: | EUPATI Open Classroom |
| Course: | Early Clinical Development |
| Book: | Early Clinical Development Plan |
| Printed by: | Guest user |
| Date: | Monday, 20 July 2026, 11:20 PM |
1. Introduction
(This section is organised in the form of a book, please follow the blue arrows to navigate through the book or by following the navigation panel on the right side of the page.)
It is important for a rational development of medicines that researchers ask relevant questions and answer them with appropriate studies. Clinical studies can be classified based on when the studies occur during clinical development (Phase I, II, III, IV) or why they are done (human pharmacology, therapeutic exploratory, therapeutic confirmatory, therapeutic use).
Before any clinical trial is carried out, results of non-clinical studies should be acceptable and show that the medicine is safe for the proposed human testing. The safety of participants in the first human studies (first-in-human studies) is the most important consideration. Non-clinical testing might find potential risk factors for investigational medicinal products (IMP). However, the ability of non-clinical studies to predict safety issues in humans may be limited because the nature of the target is more specific to humans or because of other factors. Attention should be given to the estimation of the initial dose to be used in humans and when the dose is escalated (increased).
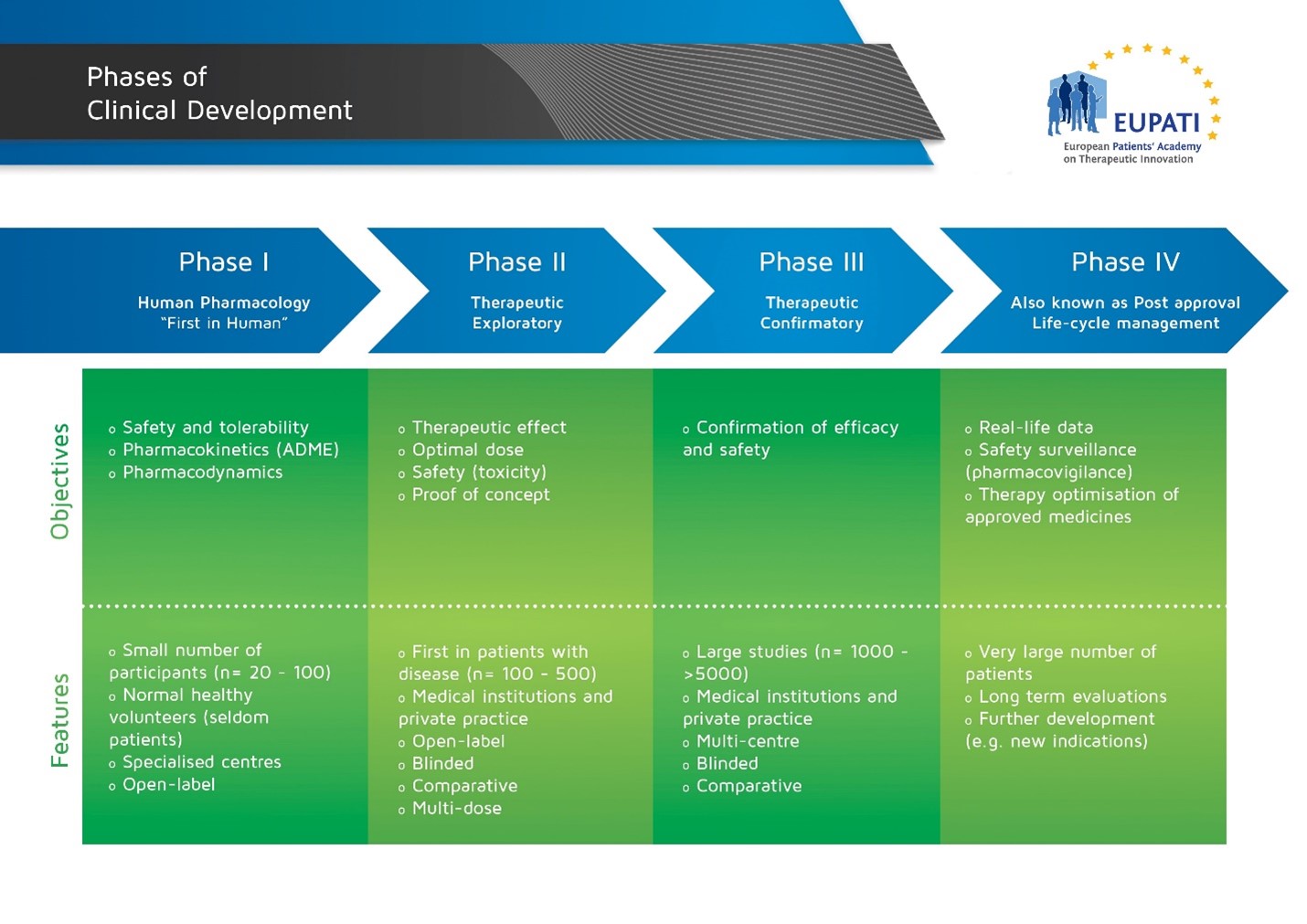
Image with details of the four phases of Clinical Development.
Studies are executed in phases in the development of a medicinal product so that the results of prior studies can influence the plan of later studies. However, the phases do not mean that the order of studies is fixed. Data that come forth during development will often prompt a change of the development strategy. New data may also suggest the need for additional studies that are typically part of an earlier phase. Below are a couple of examples.
Human pharmacology studies are usually done in Phase I but many Phase I studies are also carried out in phase II, III and IV (e.g. blood level data in a late study may suggest the need for a medicine-medicine interaction study, or adverse effects may suggest the need for further dose finding).
Phase I trials normally initiate clinical medicine development. However, a study may combine several phases with different fundamental objectives. Therefore, studies are often labeled not just as phase I, for instance, but alternatively: early Phase I (IA) or late Phase I (IB), or perhaps Phase I/II, early Phase II (IIA) or late Phase II (IIB), or Phase II/III, etc.
There are key questions that must be answered by the studies in early clinical development (Phases I & II):
Phase I
- Is the medicine safe in humans? At what levels? (Tolerance)
- What does the body do to the medicine? (Pharmacokinetics (PK))
- What does the medicine do to the body? (Pharmacodynamics (PD))
- What interactions are there? (with other medicines, substances, food and drink)
Phase II
- Is the medicine safe in patients?
- What does the medicine do to the body? (Pharmacodynamics (PD))
- Does the medicine seem to work in patients? At what dose? (Effect)
2. Key Steps in Early Clinical Development
These are the non-clinical studies that support the start of clinical studies.
- Establish programme objectives, Proof of Concept (PoC) requirements, and development plan.
- Design and conduct clinical studies: Phase I, PoC, (Phase II) (establish PK and PD assays).
- Data driven development decisions.
3. Importance of Pharmacokinetics (PK) and Pharmacodynamics (PD)
- Pharmacokinetics (PK) study what the body does to the medicine. This means they:
- Determine the plasma time-concentrations (active substance, metabolites).
- Establish the exposures that have an effect in vivo (Latin for ‘in the living’).
- Allow systematic exposure in order to assess toxicology
- Pharmacodynamics (PD) study what the medicine does to the body. This means they:
- Provide measures of on-target medicine effect and duration of effect.
- Assay depends on mechanism of action.
PK and PD influence how the dose is selected and they help schedule how it is used.
4. Types of Study and Their Objectives
| Clinical Phase |
Type of Study |
Objective of Study |
Examples |
|---|---|---|---|
| Phase I |
Human Pharmacology - Pharmacokinetics and Tolerability |
|
|
| Phase II |
Therapeutic Exploratory - Effect |
|
|
5. Timing of Human Pharmacology Studies
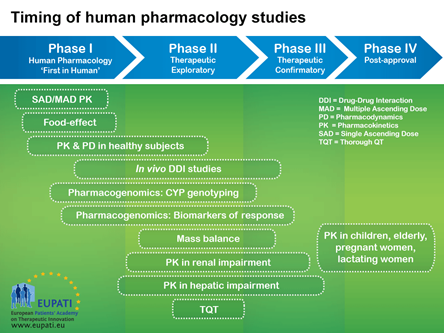
Image with details of the timing of human pharmacology studies across the four phases of medicines development.
6. Applicable regulatory guidelines (optional)
The EMA provides a number of guidelines on Non-Clinical and Clinical development.
The main guidelines related to early clinical development are the following:
Non-clinical
- Non-Clinical Safety Studies For The Conduct Of Human Clinical Trials For Pharmaceuticals (ICH M3, CPMP/ICH/286/95)
ICH M3 (R2) Non-clinical safety studies for the conduct of human clinical trials for pharmaceuticals - EMA - Preclinical Safety Evaluation of Biotechnology-derived Pharmaceuticals (ICH S6, CPMP/ICH/302/95)
ICH S6 (R1) Preclinical safety evaluation of biotechnology-derived pharmaceuticals - EMA - Non-clinical Evaluation of QT Interval Prolongation (ICH S7B, CPMP/ICH/423/02)
ICH S7B - Non-clinical Evaluation of the Potential for delayed Ventricular Repolarization (QT Interval Prolongation) - EMA - Safety Pharmacology Studies for Human Pharmaceuticals (ICH S7A, CPMP/ICH/539/00)
ICH S7A Safety pharmacology studies for human pharmaceuticals - EMA - Toxicokinetics: Assessment of Systemic Exposure in Toxicity Studies (ICH S3A, CPMP/ICH/384/95)
ICH S3A Toxicokinetics - EMA - Position Paper on Non-clinical Safety Studies to Support Clinical Trials with a Single Microdose (CPMP/SWP/2599/02)
Position Paper - IAA AMS
Clinical
- General Considerations for Clinical Trials (ICH E8, CPMP/ICH/291/95)
ICH E8 General considerations for clinical studies - EMA - Guideline on Strategies to Identify and Mitigate Risks for First-in-Human Clinical Trials (EMEA/CHMP/SWP/28367/07)
Guideline on strategies to identify and mitigate risks - EMA - Guideline for Good Clinical Practice (ICH E6, E6(R2), E6(R3))
First adopted by the EU in 1996 and most recently updated in 2025.
ICH_E6(R3)_Step4_FinalGuideline_2025_0106.pdf - Guideline on Dose Response Information to Support Drug Registration (ICH E4, CPMP/ICH/378/95)
ICH E4 Dose response information - EMA - Guideline on Studies in Support of Special Populations: Geriatrics (ICH E7, CPMP/ICH/379/95)
ICH E7 Studies in support of special populations: geriatrics - EMA - Guideline on PK of Medicinal Products in Patients with Impaired Hepatic Function (CPMP/EWP/2339/02)
Evaluation of pharmacokinetics in hepatic impairment - EMA - Guideline on PK of Medicinal Products in Patients with Decreased Renal Function (draft, EMA/83874/2014)