Key Principles of Pharmacology
| Site: | EUPATI Open Classroom |
| Course: | Early Clinical Development |
| Book: | Key Principles of Pharmacology |
| Printed by: | Guest user |
| Date: | Sunday, 12 July 2026, 1:01 PM |
1. Introduction
(This section is organised in the form of a book, please follow the blue arrows to navigate through the book or by following the navigation panel on the right side of the page.)
Pharmacology is the study of how a medicine works, how the body responds to it, and the changes that occur over time. The two main areas of pharmacology are: pharmacokinetics and pharmacodynamics. These are explained in more detail below. Non-clinical pharmacological studies allow scientists to compare a medicine’s beneficial effects with its negative (toxic) effects. This comparison is important so that a thorough benefit-risk analysis can be made before proceeding to test the medicine in human studies. If the medicine does proceed to the clinical phase, data gathered during non-clinical pharmacology and toxicology studies help to determine the dosage of medicine given to volunteers in the first- in-human studies.An overview display of the different components, playing a role in a medicines effect in the body, is given in the following graphs:
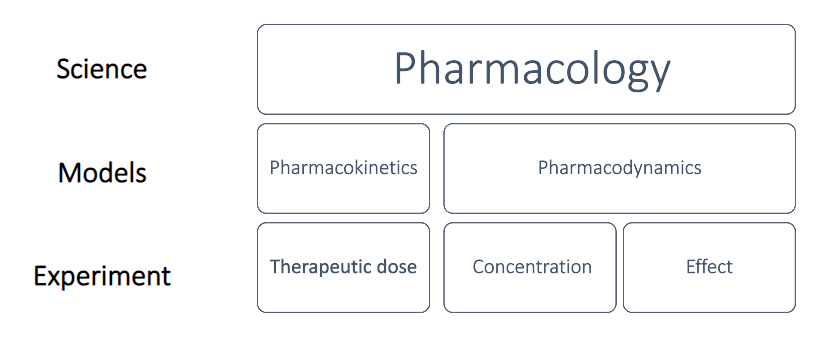
Figure.1: Schematic representation of the different components acting in human pharmacology and their classification. (adapted from ‘Introduction to Pharmacokinetics’, J. Scott Daniels, Vanderbilt Univ Medical Center, 2012).
When determining the effect a medicine has, the time course from administration to excretion plays an important role as does the interaction between dose administered, pharmacokinetics (influence on concentration) and pharmacodynamics (influence on the effect). The following schematic ‘From PK to PD’ depicts this interrelation.

Figure. 2: Schematic quantitative representation of the interaction between PK – PD – time – effect after administration of a medicine (adapted from ‘Introduction to Pharmacokinetics’,
J. Scott Daniels, Vanderbilt Univ Medical Center, 2012)
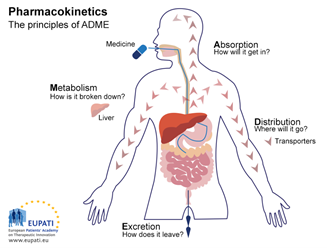
2. Pharmacokinetics
In other words, pharmacokinetics studies four major mechanisms usually identified in many text books with the acronym ADME:
- Absorption: How the medicine gets into the body
- Distribution: Where the medicine goes in the body
- Metabolism: How the body chemically modifies the medicine
- Excretion: How the body eliminates the medicine
2.1. Absorption
- Oral administration (PO: per os – by mouth) is a route where a substance (e.g. tablet, syrup) is taken through the mouth to be absorbed from the stomach or intestine, enters the liver and the general circulation. This method is used in 80% of the medicines used in medical practice because the route is convenient and economical.
- Sublingual (SL), where the medicine is dissolved under the tongue (e.g. sublingual tablets). and is absorbed through mucous membranes into the bloodstream. This route is fast and convenient.
- Transdermal (TD): medicine is contained in a patch, and absorbed through the skin (e.g. nicotine, oestrogen). The major advantage for this route is the medicine dosage is continuous and long-acting; however, it may cause local irritation due to the adhesive patch on the skin.
- Rectal (PR), where medicine (e.g. analgesic and anti-nausea suppositories) is inserted into the rectum; the medicine is absorbed through mucosa into the bloodstream. It is useful for unconscious or vomiting patients, but the medicine may be incompletely or irregularly absorbed.
- Inhalation: medicine is inhaled as a gas, aerosol or mist (e.g. bronchodilator). The medicine is usually intended to act directly on lung tissue. This route produces a rapid onset of action, but the inhaled medicine may irritate lung tissue.
- Intranasal: medicine is absorbed through the mucosa membranes in the nose into the bloodstream (e.g. anti-congestion spray).
- Intravenous - IV, intramuscular - IM, subcutaneous – SQ or SC (all of these are known as parenteral). This route produces a faster response than oral or rectal routes and avoids unpredictable absorption through the gastrointestinal system; however, it requires sterile conditions to prevent infections.
2.2. Distribution
2.3. Metabolism (medicine biotransformation)
2.4. Excretion
It refers to how the body gets rid of a medicine or its metabolites. There are three main routes of elimination: kidneys (urine), liver, and bowel (bile in faeces). Most medicines are excreted as active, partially active, or inactive metabolites by the kidneys. Infants and elderly have underdeveloped or impaired abilities to metabolise and excrete less of a medicine which usually results in greater sensitivity to medicines.3. Measuring Pharmacokinetics
Based on data gathered during pharmacokinetic studies, Pharmacokinetics (PK), applying scientific and mathematical models helps to predict and understand the time course of Absorption, Distribution, Metabolism, and Excretion of medicines in the body.. This allows scientists to assess the relationship between the medicine’s beneficial and toxic effects, and to predict the safety and tolerability of the medicine in humans. Data gathered during pharmacokinetic studies are thus essential for determining dosing schedules in clinical trials. A favourable PK profile is vital to the therapeutic success of a medicine.
Pre-clinical outcomes from doing PK are:
Select compounds that have the maximum potential of reaching the target (PK)
Select the appropriate route of administration to deliver the medicine
Understand how the blood levels relate to efficacy (PK-PD) or toxicity (TK-TD) in order to select safe doses
Decide on the frequency and duration of dosing in order to sustain medicine at target for disease modification
Predict Human pharmacokinetics
To measure the timeline of the medicine through the body, there are a few measurements that need to be undertaken and calculated:
The speed at which the medicine appears in the blood (plasma) and its concentration after administration are measured (absorption and distribution).
How the medicine changes over time (metabolism) and leaves the body (excretion) is measured.
How fast the medicine disappears as it is converted into metabolites and even secondary metabolites is calculated.
The obtained values are plotted in a chart like the one shown in Figure 4.

Figure. 4: Concentration of medicine within blood plasma (Y axis) vs. time (X axis)
If we give a person a medicine at time zero, we will see the concentration of the medicine go up and then fall slowly as it is eliminated from the body. In the first phase the medicine enters the body faster than it is being removed (absorption phase), and its concentration in the body increases to reach a maximum (Cmax.) at a certain time (Tmax.). Then, after the peak concentration has been reached, the medicine is eliminated from the body faster than it is entering, so the concentration of the medicine in the body starts decreasing (elimination phase).
By plotting plasma concentrations of the medicine versus time, we can calculate the area under the curve (AUC (from time zero to infinity), expressed in units of μg x h/mL (μg × h/mL). Cmin in the graph may refer to the detection limit of the medicine, that is the minimum concentration measurable. Whenever the determination of AUC is partial (incomplete), the time period over which it is determined should be specified, for example, AUC0–12 h refers to area under the curve from time zero to 12h after administration.
One further important parameter is the elimination half-life of a medicine, defined as the time it takes for the concentration of the medicine in the plasma or the total amount in the body to be reduced by 50% (T1/2 in the graph). In other words, after one half-life, the concentration of the medicine in the body will be half of the starting dose. With each additional half-life, proportionally less of the medicine is eliminated. However, the time required to reach half of the original concentration remains constant. In general, the medicine is considered to have a negligible therapeutic effect after 4 half-lives, that is, when only 6.25% of the original dose remains in the body. The elimination half-life is a useful pharmacokinetic parameter as it provides an accurate indication of the length of time that the effect of the medicine persists in an individual. It can also show if accumulation of the medicine is likely to occur with a multiple dosing regimen. This is helpful when it comes to deciding the appropriate dose amount and frequency. Along with other pharmacokinetic data and values about the individual patient, the half-life can help health practitioners to estimate the rate at which a drug will be eliminated from the body, and how much will remain after a given time period.
The area under the curve (AUC) shows the total exposure to a medicine that the body receives (the shading under the graph). This is a function of how high the concentration of the medicine goes and how quickly it is excreted.
3.1. Bioavailability
In order to understand and interpret pharmacokinetik measurements the concept of bioavailabity should be known. To note: Absorption and bioavailability are related concepts.
In Pharmacokinetics Made Easy (Birkett, 2009) defines bioavailability as:
"Bioavailability is the fraction of the dose which reaches systemic circulation as intact drug"
Other definitions are possible and perhaps even desirable. The flaw of most definitions is the tendency to ignore the possibility that drugs might get absorbed by other routes, and that serum concentration may not be the concentration of highest interest. For instance, the United States FDA defines bioavailability as "the rate and extent to which the active drug ingredient or therapeutic moiety is absorbed from a drug product and becomes available at the site of drug action" and the European EMA simply defines it as “The extent to which an active ingredient is absorbed from a medicine and becomes available in the body”.
Determination of bioavailability
A few points upfront:
Bioavailabilty, the fraction of the administered dose which reaches the systemic circulation unchanged, is denoted by the letter f and, if expressed as percent (which is usually the case), by the letter F
Calculation is based on the assumption that there is a direct relationship between the concentration of drug in blood or plasma and the concentration of drug at the site of action. However, consider the possibility that circulating drug levels may misrepresent its availability to its target. (Example: as the only biologically active form of the medicine phenytoin is the free drug, the circulating protein-bound fraction (up to 90%) is not available to its site of action in the brain, even though it might be well-absorbed)
When a medicine is given orally, only part of the administered dose appears in the plasma.(Example: if 100 mg of a medicine are administered orally and 70 mg of this medicine are absorbed unchanged, the bioavailability is 0.7 or seventy percent)
Various factors may affect bioavailability, such as Pharmaceutical factors, Patient related factors, Route of administration, metabolism
The most common types of human bioavailability studies are Plasma Level - Time Studies. Following the administration of a single dose of a medication, blood samples are drawn at specific time intervals and analysed for drug content.
Bioavailability is then calculated by comparing plasma levels of a drug given via a particular route of administration (for example, orally) with plasma drug levels achieved by IV injection. However, the concentration of a drug given IV will be maximal at Time Zero, whereas the concentration of an orally administered drug will be maximal at a later time (depending on the rate of absorption). This is where the AUC comes into play (the area under the curve calculated by plotting plasma concentrations of the drug versus time). The AUC equals the total exposure to an API that the body receives. In brief, the mathematical concept of Dost's Law of Corresponding Areas (1972) relates the AUC of an IV dose to the AUC of an orally administered dose. If the area under the oral concentration curve covers 50% of the area covered by the IV curve, the law dictates that the drug is 50% bioavailable.
The bioavailability of the intravenous dose of any drug is by definition 100%. Hence, the bioavailability of all other formulations and routes of administration can be compared to this reference value as an absolute standard, and from this the equation for absolute bioavailability can be derived:
The absolute bioavailability then is the dose-corrected area under the curve (AUC) non-intravenous divided by AUC intravenous. The equation for calculating the absolute bioavailability (denoted by the letter f or, if expressed in percent, by F), of a drug administered orally (tablet) is given below:

Bioavailability of a drug administered orally is the ratio of the area calculated for oral administration compared with the area calculated for IV injection.
Whereas absolute bioavailability compares the drug formulation to an IV dose, relative bioavailability compares it to another similar non-IV formulation. One may be comparing two tablets, or a tablet to a suppository, or something similar.
A graphic representation of the associated plots for an IV and an oral drug administration is given in the following figure.

Fig. 5: Concentration vs. time graph of AUC of IV and oral administration of a medicine
In summary:
Bioavailability is the fraction of the dose which reaches systemic circulation intact
IV bioavailability is by definition 100%
"Absolute" bioavailability compares one non-IV route with IV administration.
"Relative" bioavailability compares one non-IV route or formulation with another (instead of using IV route as a reference).
3.2. Bioequivalence
Birkett (2009) describes bioequivalence as "a clinical definition referring to two formulations of a drug".
"Drugs are considered bioequivalent if the extents and rates of absorption of drug from them are so similar that there is likely no clinically important difference between their effects"
The EMA states “Two medicinal products containing the same active substance are considered bioequivalent if they are pharmaceutically equivalent or pharmaceutical alternatives and their bioavailabilities (rate and extent) after administration in the same molar dose lie within acceptable predefined limits. These limits are set to ensure comparable in vivo performance, i.e. similarity in terms of safety and efficacy”.
Bioequivalence rests on the assumption that the measured medicine concentration is related to its clinical effect. If the active ingredient from both drug formulations achieves the same concentration at the same rate (or AUC of the concentration over time curve) then likely these two medicines will achieve the same clinical effect, and so they can be considered bioequivalent.
If one is able to agree on what one views as a clinically relevant effect, one may be able to test bioequivalence empirically. Typically one takes the serum concentration as the easily measurable and clinically relevant parameter. The two medicines are then compared in terms of their relative bioavailability. If the AUC ratio of the two medicines is within the range of 0.8-1.25, the claim can be made that the two formulations are bioequivalent (Rescigno, 1992) and a generic competitor product could obtain a marketing authorisation.
4. Pharmacodynamics
Pharmacodynamics (PD) is the study of the effect of a medicine on the body.

There are two ways a medicine can affect the body:
 A medicine can change conditions within the body, or
A medicine can change conditions within the body, or
A medicine can interact with specific parts of the body on a cellular or sub-cellular level.
The primary objective of pharmacodynamic studies is to gather information on how the medicine affects the body this can be biochemical, physiologic, and molecular effects and involves receptor binding (including receptor sensitivity), postreceptor effects, and chemical interactions).). It allows scientists to assess the efficacy of the medicine – that is, whether or not the medicine is having the desired effect on the target, and if so, how strong that effect is.
It also allows a better understanding of the relationship between the concentration of the medicine in the body and the strength of its effect. This means that PK and PD both together influence dosing, benefit, and adverse effects (for example, addressed in PK/PD models).
The majority of medicines either mimic or inhibit normal physiological/biochemical processes or inhibit pathological processes.
There are 7 main medicine actions:
stimulating action through direct receptor agonism and downstream effects
depressing action through direct receptor agonism and downstream effects
blocking/antagonizing action, the medicine binds the receptor but does not activate it
stabilizing action, the medicine seems to act neither as a stimulant or as a depressant
exchanging/replacing substances or accumulating them to form a reserve
direct beneficial chemical reaction as in free radical scavenging
direct harmful chemical reaction which might result in damage or destruction of the cells, through induced toxic or lethal damage
Pharmacodynamics places particular emphasis on dose–response relationships, In principle, then the goal would be to dose for an optimal plasma concentration of the medicine for a desired level of response. In reality, there are many factors affecting this goal. Pharmacokinetic factors determine peak concentrations, and concentrations cannot be maintained with absolute consistency because of metabolic breakdown and excretory clearance. Genetic factors may exist which would alter metabolism or drug action itself, and a patient's immediate status may also affect indicated dosage.
A simplified schematic of PK and PD parameters and their interrelation is given in the following figure (Fig. 6).

Figure.6: Relationship between the plasma concentration – time curve obtained following a single extravascular (e.g. oral) dose of a medicine and parameters associated with the therapeutic or pharmacological response
Two parameters, as shown in the above figure, deserve specific mention because they give an indication of the efficacy and safety of the medication tested:
The therapeutic window is the amount of a medication between the amount that gives an effect (minimum effective concentration) and the amount that gives more adverse effects than desired effects (maximum safe concentration). To note: adverse effects (side effects) of various nature can occur while the medicine is effective (benefit) but their influence (risk) must be less than the positive effect (a favourable benefit – risk ratio) over the range of intensity (from minimum effective concentration to Cmax) to make the medicine acceptable. Therefore, medication with a small therapeutic window must be administered with care and control, e.g. by frequently measuring blood concentration of the drug.
The duration of action of a medicine is the length of time that the particular drug is effective (from reaching the minimum effective concentration in the absorption phase back to this level in the elimination phase). Duration of action is a function of several parameters including plasma half-life, the time to equilibrate between plasma and target (compartments), and the off rate of the drug from its biological target.
5. Further Reading
- Guideline on Strategies to Identify and Mitigate Risks in First-in-Human Clinical Trials with investigational Medicinal Products Available at: Guideline on Strategies to Identify and Mitigate Risks for First-In-Human Clinical Trials with Investigational Medicinal Products (europa.eu)
- Non-Clinical Safety Studies for the Conduct of Human Clinical Trials for Pharmaceuticals: Available at: ICH M3 (R2): Non-Clinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals (europa.eu)
- Preclinical safety evaluation of biotechnology-derived pharmaceuticals: Available at: S6(R1) Step 5 Preclinical safety evaluation of biotechnology-derived pharmaceuticals (europa.eu)
- The Non-Clinical Evaluation of the Potential for delayed Ventricular Repolarisation (QT Interval Prolongation) by Human Pharmaceuticals: Available at: 05-20959.pdf (govinfo.gov)
- Safety pharmacology studies for human pharmaceuticals: Available at: S 7 A Safety Pharmacology Studies for Human Pharmaceuticals (europa.eu)
- Toxicokinetics: the assessment of systemic exposure in toxicology studies: Available at: S 3 A Toxicokinetics: A Guidance for Assessing Systemic Exposure in Toxicology Studies (europa.eu)
- Position paper on the non-clinical safety studies to support clinical trials with a single micro dose: Available at: M3(R2) Step 5 Non-clinical safety studies for conduct of human clinical trials for pharmaceuticals (europa.eu)
- Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers: Available at: Guidance for Industry (fda.gov)