Principles of New Trial Designs and their Practical Implications
| Site: | EUPATI Open Classroom |
| Course: | Clinical Trials and Trial Management |
| Book: | Principles of New Trial Designs and their Practical Implications |
| Printed by: | Guest user |
| Date: | Saturday, 18 July 2026, 3:53 PM |
1. Introduction
(This section is organised in the form of a book, please follow the blue arrows to navigate through the book or by following the navigation panel on the right side of the page.)
In the traditional paradigm of clinical trial design, each new treatment must go through a strict development process. After successful Phase I trials, a Phase II trial is needed to show sufficient efficacy and safety. When this has been demonstrated, the medicine goes into Phase III trials, where it is compared with a standard treatment (control). Doing this for each treatment separately requires a long period of time, a large number of patients, and substantial financial resources. Additionally, in the traditional approach, modifications are not allowed during the course of the trial.
One new approach to clinical trial design is an adaptive clinical trial design. Adaptive clinical trials include a pre-planned opportunity for modification of one or more specified aspects of the trial. This is usually based on the analysis of interim data from participants during the trial.
There are many reasons to use adaptive designs (or adaptive pathways) in clinical trials. In an environment subject to economic challenges, adaptive designs appear to be appealing for pharmaceutical industry, academic institutions, clinicians, and patients.
2. Adaptive Designs
Adaptive designs are relatively flexible clinical trial designs, allowing for modifications during the course of the trial in order to streamline and optimise the process. Analyses of the accumulating study data are performed at pre-planned time points within the trial (interim analysis), can be performed in a fully blinded or unblinded manner, and can occur with or without formal statistical hypothesis testing. Adaptive designs allow for real-time learning during the course of a trial; they are relatively flexible as they allow for modifications during the course of the trial, in order to streamline and optimise the process. It is important that the process is modified only in such a way that the validity and integrity of the trial are not affected.
Adaptive designs can pose operational challenges because of their complexity, and the process of adapting a trial can introduce bias. This bias can be either statistical or operational – for example, if an adaptation suggests that the results of a trial point in a certain direction.
The adaptive design may improve trial efficiency for the sponsor and the participants in the trial. However, if it is not properly conducted, there is a high risk that such a trial can generate clinical results that are difficult to interpret or translate into daily practice.
2.1. Adaptive Designs in Rare Diseases
Clinical trials for rare diseases are typically small out of necessity. Planning a small clinical trial, particularly for a rare disease, can present several challenges. Small trials exhibit more variability than larger trials, which implies that standard designs may lead to trials adequate only for large effects.
The specific requirements of rare disease trials make adaptive designs particularly appealing. Classical trials for rare disease are typically powered for large effects. The power of a statistical test is the ability of the test to detect an effect, if the effect actually exists. In statistical terms, it is the probability that it will correctly lead to the rejection of a null hypothesis.
In some cases we may not be able to reject the null hypothesis, not because it is true, but because we do not have sufficient evidence against it. This might be because the experiment is not large enough to reject the null hypothesis. As such, the power of a test can be described as the probability of not making a Type II error (not rejecting the null hypothesis when in fact it is false).
Adaptive designs provide an appealing alternative because:
- They shorten the development process without compromising validity or efficacy.
- Ineffective treatments can be identified earlier on.
- They permit a more efficient use of resources.
3. Possible Approaches in Adaptive Design
The term ‘adaptive’ covers a varied set of designs, but most of them follow a simple structure. Within an adaptive trial, there are learning and confirming stages, which follow a similar approach to the overall clinical development process across multiple trial settings (Phase I, Phase II, and Phase III). As a result, changes might be made to hypotheses or the design parameters.
Learning stages
- Major design elements may be changed (for instance dropping treatment arms).
- Statistical uncertainty (for instance bias, variability, incorrect selection).
- Estimation of the treatment effects (beneficial or adverse).
How the ‘learning’ part is done is crucial to the integrity and the validity of the trial results. Modifications based on blinded interim results will have less impact on the trial operating characteristics. However, adaptations based on unblinded interim comparative analyses can introduce all sorts of bias.
Confirming stages
- Control of statistical errors and operational biases are of utmost importance.
- Strong control of Type I errors is required (for example, finding a treatment efficient when it, in fact, is not).
These rules are predetermined and are verified by one or more interim analyses. They prevent the participants from taking medicines that will not provide a beneficial effect or are unsafe. Most importantly, if it is found that the trial medicine is clinically more effective than the control, it would be unethical to continue administering the less-effective control medicine. Early stopping rules for futility allow a halt in the administration of a less-effective control medicine.
There are also designs where treatment arms are modified or dropped over the course of a trial, or where a sub-population is selected based on a biomarker of interest (´pick the winner´).
Some designs allow for sample size re-estimation, for instance an increase in the patient population if the results appear promising; or to maintain overall statistical power. At the interim analysis, a check is done for efficacy or futility. At this point the trial can be stopped in the presence of overwhelming evidence of efficacy or futility. If not, the conditional power (CP) is determined. The CP is the probability that the final study results will be statistically significant given the data currently observed. If this CP is either high or low then the trial is continued as planned. If the CP is somewhere in the middle, then the sample size is further increased.
During trial recruitment, if the expected number of participants under the original eligibility criteria cannot be enrolled, modifications to non-critical eligibility criteria can be made based examination of baseline characteristics.
Adaptive randomisation is another example of an intuitively appealing design. In this design, a higher proportion of patients would be treated with the ‘better’ arm (if there is one). These adaptive trial designs are mostly based on unblended interim analyses that estimate the treatment effects – meaning that the analysts are aware of which treatment participants have been allocated to.
3.1. Example 1: Group Sequential Design
A group sequential design is a typical example of a Phase III trial with rules for early stopping for futility or efficacy. In the example trial depicted in the diagram below, patients were randomised between the first line treatment with either one medicine alone or two medicines in combination.
There were two interim stages where it was possible to stop the trial early and performing analysis before all the trial results are gathered. The trial could have been stopped:
- At Interim 1, for futility based on progression-free survival (PFS) – whether the patient stays free of any progression of a specific cancer or not.
- At Interim 2, for futility or efficacy based on overall survival.
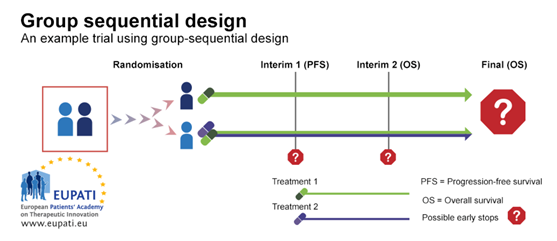
Group
sequential design
Group sequential design allows for early stops on the basis of progression-free
survival or overall survival. In this example, participants were randomised
onto one of two arms, and received either Treatment 1, or a combination of
Treatment 1 and Treatment 2.
3.2. Example 2: Multi-arm, Multi-stage Design (MAMS)
The multi-arm, multi stage (MAMS) trial is a new paradigm for conducting randomised controlled trials which makes use of an interesting adaptive design.
MAMS trials allow the simultaneous assessment of a number of research treatments against a single control arm. MAMS trials provide earlier answers and are potentially more cost-effective than a series of traditionally designed trials.
In this example, we see a design that uses multiple arms and stages at the same time.
The MAMS design requires a definitive primary and intermediate primary outcome measure. The definitive outcome measure is the one upon which the final conclusions should be based; the intermediate outcome measure provides a means of screening for emerging
evidence of evidence.
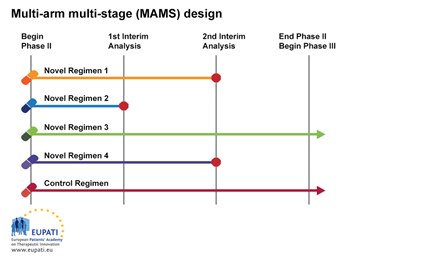
Multi-arm multi-stage design
The multi-arm multi-stage design (MAMS) allows multiple treatments to be tested simultaneously against a single control.
At the first interim analysis in the example above, Novel Regimen 2 is considered to lack sufficient benefit compared with the control and is not taken forward to stage 2. At the second interim analysis, recruitment to Novel Regimens 1 and 4 is stopped, and only the control regimen and Novel Regimen 3 are continued to the end of trial and advanced into Phase III studies.
Advantages of the MAMS design
- Fewer participants
In this design, several trials are performed at once, which helps reduce the number of participants randomised to the control arm. - Less overall time required for medicine discovery
The intermediate steps of the MAMS design replace the separate Phase II step. The decision on whether the medicine is sufficiently active is incorporated as a pilot phase into this trial. - Fewer applications and approvals required
Regulatory work is done for one trial instead of for multiple trials. - Flexible
Uninteresting arms can be dropped and new arms can be added. - Reduced cost
This trial design requires fewer participants, fewer regulatory applications, and less overall time, all of which help to save on development costs.
Disadvantages in MAMS design
- Operating characteristics
Because of the complexity of this approach, it may be difficult to manage and requires a lot of simulations during the design process. - Required number of participants
This depends on the operating characteristics, but if treatment arms are added during the course of the trial, it may be difficult to predict budget and regulatory issues. - Trial duration
If treatment arms are added, it becomes difficult to predict when the trial will naturally end. - Continued accrual (recruitment) to control arm
In order to avoid a time bias when new treatment arms are added, recruitment to the control arm must continue throughout the course of the trial. Consideration must also be given to what happens if a new standard of care becomes available during the course of the trial – is the control still relevant? - Comparison between experimental arms
The MAMS design only allows for comparisons between individual treatment arms and the control arm; it does not allow for comparison between individual treatment arms themselves.
3.3. Example 3: Seamless Phase II/Phase III Design
Seamless Phase II/Phase III design is often used in the case of rare diseases; it is also called a ‘combination test’. In the example below, patients are randomised between three treatment arms in the first stage of the design (Phase IIb). The first treatment arm is the control arm, where patients receive the standard of care therapy. Patients on the second and third treatment arms receive different treatments, Treatment 1 or Treatment 2.
At the end of the first stage (Phase IIb), Treatment 1 and Treatment 2 are compared based on best progression-free survival (PFS). The least effective treatment arm is dropped. The other treatment arm is then continued in the second stage (Phase III). In this stage, an efficacy comparison is performed against the standard of care treatment.
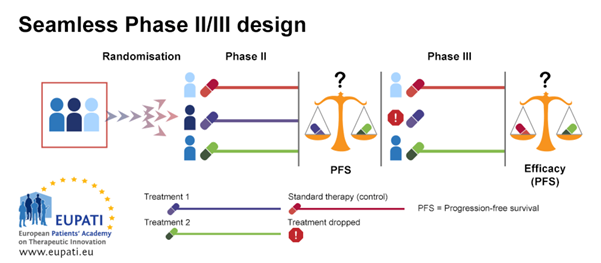
Seamless
Phase II/III design
The seamless Phase II/III design allows Phase II and Phase III to be performed
in the context of one trial.
Advantages in Seamless Phase II/Phase III design
- Helps to mitigate bias
Both steps are conducted independently and the results of both steps are combined in the end in an overall test result.
- Shortens time and participant exposure
Phase II and Phase III are performed within the context of one trial.
-
Relatively flexible
The way that the treatment arm for final comparison is chosen in the Phase II part and merged with the Phase III part is relatively flexible.
-
Efficient use of resources
Participants from Phase II and Phase III both contribute data to the final results.
Disadvantages
- Complicated statistical analyses
This design requires statistical aspects that are not so straightforward.
- Recruitment gaps
There is a gap in the recruitment between the two phases while waiting for enough data to be gathered in order to perform the interim analysis that decides whether to continue or not.
- Logistic challenges
This design is logistically challenging – it requires a quick flow of data so that the number of events in the analysis can be followed up on.
- Difficulties arising from long-term endpoints
This design requires information on PFS to be available relatively quickly. This becomes more difficult when the endpoints are long-term.
- Risk of lost information
Combining two arms risks the loss of information.
3.4. Example 4: Responsive-Adaptive Randomisation
In this design, patients are hierarchically randomised based on their biomarker profile into one of four treatment arms. They start with a set of initial randomisation probabilities and then based on the observed eight-week outcome, new randomisation probabilities are derived. This maximises the chance that the patient receives the treatment that is most effective for him/her.
To be able to make use of response-adaptive randomisation, a few pre-requirements are necessary:
First, such a trial requires a fast dataflow. Data on the eight-week endpoint needs to be rapidly available in the data centre so that they can update the randomisation probabilities guiding new participants entering the trial. This is not always straightforward in the context of a large multicentre trial.
This example had a short endpoint (eight-week). It would not work so well if a Progression Free Survival (PFS) at six months was needed to guide randomisation of new participants.
It is difficult to interpret results beyond estimation. It is difficult to perform comparisons when no longer working with the classical concept of two independent samples. In addition, it can’t be conclusive for an arm that was prematurely terminated and for which little data is available.
When investigators start to realise that more participants are being randomised into a particular treatment arm, recruitment patterns may change during the course of a trial. This introduces operational bias, e.g. sicker participants could enrol earlier and healthier ones could decide to wait for a higher chance of receiving better treatment. In such a setting, blinding is essential but may not always be feasible.
4. Patient Involvement
Patient input into adaptive design can help researches identify the most appropriate design by helping to define and understand the needs and requirements of the patient population. Patients can also be involved in the Data Safety Monitoring Board.5. Further Reading
- Clinical Trials Planned with an Adaptive Design (2007): Available at: Reflection Paper on Methodological Issues in Confirmatory Clinical Trials Planned with an Adaptive Design (europa.eu)
- Chow SC, Chang M. Adaptive design methods in clinical trials - a review. Orphanet J Rare Dis. 3(11) May 2008: Available at: 1750-1172-3-11.pdf (biomedcentral.com)
- I. Judson, J. Verweij, H. Gelderblom, et al. Results of a randomised phase III trial (EORTC 62012) of single agent doxorubicin versus doxorubicin plus ifosfamide as first line chemotherapy for patients with advanced or metastatic soft tissue sarcoma: a survival study by the EORTC Soft Tissue and Bone Sarcoma Group.
- Sydes MR, Parmar MK, James ND, et al. Issues in applying multi-arm multi-stage methodology to a clinical trial in prostate cancer: the MRC STAMPEDE trial. Trials. 10 (39) Jun 2009: Available at: 1745-6215-10-39.fm (nih.gov)
- Kairalla JA, Coffey CS, Thomann MA, Muller KE. Adaptive trial designs: a review of barriers and opportunities. Trials. 13 (145) Aug 2012: Available at: 1745-6215-13-145.pdf (biomedcentral.com)
- Cytel Webinar for East®SurvAdapt. October 28, 2010: Available at: Survadapt-Webinar_2014_SLIDES | PPT