Study Documentation
| Site: | EUPATI Open Classroom |
| Course: | Documentation & Management |
| Book: | Study Documentation |
| Printed by: | Guest user |
| Date: | Monday, 27 July 2026, 3:13 AM |
1. What is Source Data?
(This section is organised in the form of a book, please follow the blue arrows to navigate through the book or by following the navigation panel on the right side of the page.)
The ICH-GCP Guideline defines ‘source data’ as:
‘All information in original records and certified copies of original records of clinical findings, observations, or other activities in a clinical trial necessary for the reconstruction and evaluation of the trial. Source data are contained in source documents (original records or certified copies)’.
This means that there are various types of data that are classed as source data. For example:
- Information which the investigator writes down in the patient’s record.
- Values in a lab result sheet or direct printout from a measuring device (e.g. analysis scale, spectrophotometer).
- Answers which a patient enters into a questionnaire.
2. What Are Source Documents?
Source documents contain source data. They are original documents, data and records in which these original (source) data are noted. Examples include:- Hospital records and clinical charts.
- Lab result sheets.
- Memos.
- Patients’ diaries or evaluation checklists.
- Medication dispensing records.
- Recorded data from automated instruments, ECG, X-ray.
- Informed consent form.
Copies are only recognised as ‘source documents’ if they are certified after verification as being accurate.
2.1. Example of a Source Document
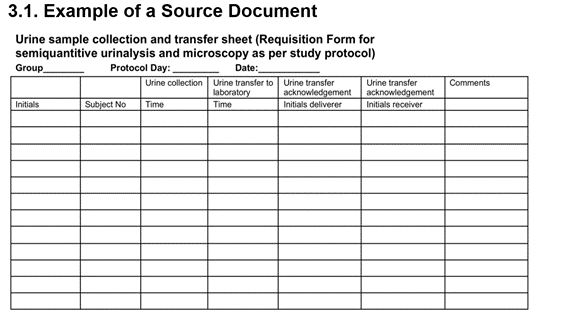
This example of a source document is a form that can be used when a study participant (subject) gives a urine sample:
- The study nurse will enter their initials, subject number, and time of urine collection. This information is noted for the first time - it is source data.
- The person transporting the urine samples to the lab will note the time of transport and sign with their initials so that it is clear who did this and when.
- The lab technician who receives the urine samples signs the form and notes the time.
2.2. Characteristics of Source Documents
Having good source documents in a clinical trial is vital for the quality of the study. Therefore they need to fulfil certain characteristics:
- Attributable - it must be clear to which participant they belong.
- Legible - they must be readable.
- Contemporaneous - they must be noted immediately after the data is generated.
- Original - they must be the original.
- Accurate - they must be reliably correct.
- Originator information – who entered the data and when.
For an inspection the source data must be made available in a computerised format compliant with the EMA Q&A: Good clinical practice (GCP). For this purpose CRFs are prepared. The investigator and study staff are requested to enter the required data from a patient’s source documents into a patient-specific CRF. It is very important that no mistakes are made in this transcription process and that the information provided in the CRFs is complete.
3. What is a Case Report Form (CRF)?
The ICH-GCP Guideline glossary defines a ‘case report form’ (CRF) as:
‘A printed, optical, or electronic document designed to record all of the protocol required information to be reported to the sponsor on each trial participant's.'
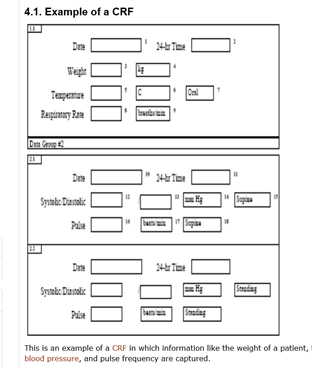
3.1. CRF Content
Here are some examples of data that are typically collected in a CRF:
- Inclusion and exclusion criteria (features a patient needs to fulfil to get enrolled into the study).
- Demographics (the patient’s personal information in an anonymised way).
- The patient’s medical history and the result of the physical examination by the doctor.
- A description of the diseases that the patient currently has and the medicine they are taking.
- Efficacy and safety parameters (measurements that show the effect of the medicine and the potential side effects the patient is experiencing).
- Which medicine, usually in a blinded fashion, was administered.
- Termination of trial (the result of the final examinations and confirmation of the patient’s completion of the study).
3.2. Regulatory Requirements for CRFs
CRFs are prepared once the protocol is agreed and before the study starts (i.e. before the first participant is enrolled). CRFs must provide the format for capturing all information that is expected to be collected in the study, as outlined in the protocol. The ICH Guidance on Statistical Principles for Clinical Trials establishes that:
- Data collected must be in full accordance with the protocol.
- CRF must be established before study starts.
- Data collected should enable the analysis to be performed.
- Identification of protocol compliance. - The CRF is also a good tool to check how well the study staff are following the protocol. If data is missing, measurement dates are not in line with the requested protocol timelines and data differ between the source documents and the CRF, this indicates that errors were made and need to be corrected.
- Participant codes must allow identification of all data reported for each participant – unambiguously.
4. Source Data Verification
The study monitor has to compare the content of the CRFs with the content of the source documents. This is called ‘source data verification’. The monitor also checks if all the required data of the participant is present in the CRF entries, and that data is consistent with the source documents.
Previously, the rule was that 100% of all CRF entries were to be compared with the source data. Nowadays, the intensity of source data verification can be reduced. The concept of ‘risk-based monitoring’ allows the sponsor to decide to which degree the monitor needs to check individual types of data. Typically, the data that is checked most thoroughly is:
- The signed Informed Consent documentation (often called Informed Consent Form – ICF).
- The measurements that are most critical for answering the study’s scientific question(s).
- The adverse events.
- The inclusion/exclusion criteria adherence.
5. Essential Documents
According to the ICH Guideline, Section 1.23, ‘essential documents’ are defined as:
‘Documents which individually and collectively permit evaluation of the conduct of a study and the quality of the data produced’.
The GCP Guideline gives under section 8 (Essential documents for the conduct of a clinical trial) a minimum list of all documents that need to be generated, filed and archived by the investigator and by the sponsor. Some of these documents need to be filed in both places like the protocol or the Investigator’s Brochure (IB). Some other documents are only to be filed by one party, for example:
The investigator files:
- The participant identification code list to document that investigator/institution keeps a confidential list of names of all participants allocated to trial numbers on enrolling in the trial. Allows investigator/institution to reveal identity of any participant.
- The signed Informed Consent documentation.
- Advertisement for participants recruitment (if used) to document that recruitment measures are appropriate and not coercive.
- Documents concerning the label(s) attached to investigational medicinal product container(s) to document compliance with applicable labelling regulations and appropriateness of instructions provided to the participants.
- Samples of the labels.
5.1. Safe-Keeping
The investigator must ensure that the ‘Investigator Site File’ (ISF) (officially the ‘Investigator Trial Master File (TMF)’) is safely archived in such a way that the documents and data can be found and read at a later time if questions come up or an inspection by the competent authorities takes place.
Systems must be in place to ensure the safe long-term storage of essential documents:
- At investigator sites, and
- by, or on behalf of, the sponsor.
6. Clinical Trial Master File (TMF)
The EMA Reflection paper on GCP compliance in relation to trial master files (paper and/or electronic) for management, audit and inspection of clinical trials (Read the full paper EMA/INS/GCP/636736/2012) defines the minimum time the sponsor and investigator need to retain the essential documents relating to a clinical trial. The duration varies by situation and member state.
Trial master files (TMF) should be established at the beginning of the trial, both at the investigator/institution site and at the sponsor's office. A TMF is the collection of documentation that allows the conduct of the clinical trial, the integrity of the trial data and the compliance of the trial with GCP to be evaluated (monitoring by the sponsor (audits) and inspection by member states). The TMF is normally composed of a sponsor TMF, held by the sponsor organisation, and an investigator TMF held by the investigator(s). The TMF kept by the investigator and that kept by the sponsor may have a different content if this is justified by the different nature of the responsibilities of the investigator and the sponsor. The sponsor needs to make sure that no documents in the TMF contain information that could be used to identify the participant.
The sponsor's TMF contains:
- The ‘study file’ - this includes all of the sponsor’s documents.
- The ‘site file’ - this is documentation that demonstrates the sponsor’s involvement in getting study authorisation for the site and overseeing the trial activities.
You can download an excerpt from the minimum list of essential documents (for a TMF) in section 8 ICH-GCP guideline. Access the complete list.
The various documents are grouped in three sections according to the stage of the trial during which they will normally be generated:
- Before the clinical phase of the trial commences,
- During the clinical conduct of the trial, and
- After completion or termination of the trial
7. Archiving
Archiving is an important means to ensure that data and documents can be retrieved at a later stage. All essential records must be appropriately archived, in conditions that include:
- Adequate and suitable space.
- Access restricted to authorised named individuals.
- Secure storage facilities with appropriate environmental controls and protection from physical damage.
Systems used for transfer of original documents to another media for storage must be validated (their reliability must be confirmed), and the availability of the equipment to convert records to readable format must be ensured.
8. Further Reading
Except from the minimum list of essential documents for Trial Master Files according to section 8 ICHGCP guideline. Taken from http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf