The Predictive Value of Non-Clinical Testing
| Site: | EUPATI Open Classroom |
| Course: | Requirements for Non-clinical Studies and the Purpose and Relevance of Animal Testing |
| Book: | The Predictive Value of Non-Clinical Testing |
| Printed by: | Guest user |
| Date: | Wednesday, 1 July 2026, 3:13 PM |
Section Overview
- 1. Introduction
- 2. From laboratory and animal studies to patients
- 3. Non-clinical studies: Ability to predict studies in humans
- 4. Timing of non-clinical studies to the clinical trial programme
- 5. Toxicology Studies
- 6. Inclusion of Women of Childbearing Potential in Clinical Trials and Embryo-Foetal Development Toxicity Studies
- 7. Setting the 'First-in-Human' Dose
1. Introduction
(This section is organised in the form of a book, please follow the blue arrows to navigate through the book or by following the navigation panel on the right side of the page.)
The data from early on in non-clinical studies help researchers make decisions on the efficacy and safety of the medicine’s development. Such decisions include the conditions and specifications of the marketing authorisation, use of the medicine on the market, pharmacovigilance (post-marketing safety monitoring), and risk management (planning and mitigation).
Non-clinical information is part of the prescription of medicines to patients, e.g., pharmacology or appropriate information for use in pregnancy and lactation. It directs the prescriber (and patients) to the optimal use of the medicine for each patient. For medicines that do not require a prescription (Over the counter, OTC), the relevant information for proper use is given in the Package Leaflet (PL). It is a challenge to give clear directions on non-clinical data in the PL.
Information gathered during non-clinical studies plays a key role in:- Decisions on clinical trials.
- Decisions on marketing authorisation applications.
- Decisions on post-marketing or monitoring studies.
- First-in-Human studies (based on pharmacology and toxicology data.
- Clinical studies specific on heart function.
- Safety monitoring in diverse systems (liver, central nervous system (CNS), renal, etc.).
2. From laboratory and animal studies to patients
A medicine candidate cannot be given to humans until enough information has been obtained on the safety and on the effects that the medicine is expected to have. Non-clinical studies bring important information that can predict the medicine’s profile, such as ‘proof of concept’, a proposed dosage regimen, and adequate safety monitoring. Also, they help define the criteria for inclusion or exclusion of patients/participants in clinical studies.
Non-clinical cell (in vitro) and animal studies (in vivo) should therefore:- Demonstrate the efficacy of the medicine candidate.
- Provide knowledge on the safety of the medicine, for instance studies that examine the maximum tolerated dose.
- Estimate the effects of the medicine that cannot be studied in humans – for instance, its effect on foetuses or in pregnant women.
Reseachers need to use their professional judgement to apply or deduce information from laboratory and animal studies to human studies. Useful rules for this so-called extrapolation have been developed and described in the guidelines of the Committee for Human Medicinal Products (CHMP) of the EMA and the ICH. These guidelines specify the types of studies that must take place before clinical trials can start. Issues with the non-clinical programme of a development candidate can cause objections when the regulatory authorities review the marketing authorisation application (MAA). Regulators may ask questions on how relevant the non-clinical models are for the proposed indication of the development candidate in humans. If researchers wish to avoid these issues, non-clinical studies must be carefully planned and laboratory and animal studies must be setup so that they can predict how the medicine will work in humans.
Researchers should plan the non-clinical programme based on the following factors:- The type of medicinal product.
- The type and severity of the disease.
- The population or group of patients that the medicine is intended for.
- The phase of the clinical trial (Phase I, II or III).
- The expected dose and duration of the treatment in humans.
Many companies seek scientific advice from regulatory authorities (for instance the EMA or national competent authorities (NCAs)). Scientific advice helps the company to make sure that the right tests and studies are done. It will also help avoid any major objections on the design of the tests later when the regulatory authorities evaluate the MAA. If companies seek and follow advice from regulatory agencies, it is more likely that they will obtain a positive outcome for an MAA. The advice is given in the light of the current scientific knowledge, and is based on the documentation provided by the company.
Non-clinical data are most important in the early development of a development candidate (see Figure 1 below). Much of the non-clinical data on safety and efficacy will be replaced later with data from clinical trials in humans and when the medicine is ready to be prescribed to patients. However, in some cases the clinical use of a new medicine will be based on non-clinical data for a longer period of time. This is due to ethical concerns about testing in humans. For example when testing the effect of a medicinal product on cancer development or in reproduction. Eventually these non-clinical data will also need to be replaced e.g. as part of post-marketing life cycle management and pharmacovigilance.
Ideally, the company should make sure that all non-clinical safety concerns are raised in the development period and before the MAA. However, there may still be safety concerns when the MAA is submitted and assessed. Such concerns can include carcinogenicity, genotoxicity, genotoxic impurities, reproductive toxicity, and hepatoxicity.
When the regulatory authorities evaluate the non-clinical programme, they may also ask the company to justify animal models. And they may question if toxicokinetics studies are sufficient to assess safety in humans.
2.1. Ethical Considerations
The Declaration of Helsinki has defined when it is acceptable to use animal models to assess the risk in humans and to mimic human diseases. It also defines when a development candidate can be given to healthy volunteers from an ethical and scientific point of view. The declaration states that biomedical research should be based on laboratory and animal experiments that are adequately done. Also, the research should be based on deep knowledge of the scientific literature. Researchers must respect the welfare of animals used in the research.
Figures 1 and 2 show how important non-clinical studies are related to clinical trial data during the medicines development process. As shown in Figure 1, researchers rely most on data from the general toxicology studies until late in clinical development. These studies monitor target organs and establish adequate safety margins of exposure. When the medicine is available for marketing, most of the non-clinical data will then be replaced by data from clinical trials.
However, for some aspects of adverse effects, such as a medicines influence on cancer development or in reproduction, it is not ethical to perform the studies directly in humans. In such situations, the clinical use of new medicines might be directed by the non-clinical data for longer periods after marketing approval. This is shown in Figure 2.
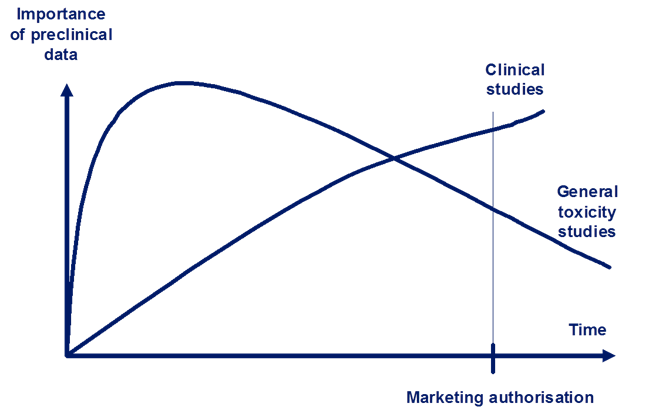
Figure 1: The relative importance and reliance of non-clinical data for human safety. Data from general toxicity studies are relied upon more than clinical data until late in the development process (intersection of the two lines).
Adapted from Non-clinical Assessment Requirements. Maria Nieto-Gutierrez; Safety and Efficacy of Medicines/Human Medicines Development and Evaluation.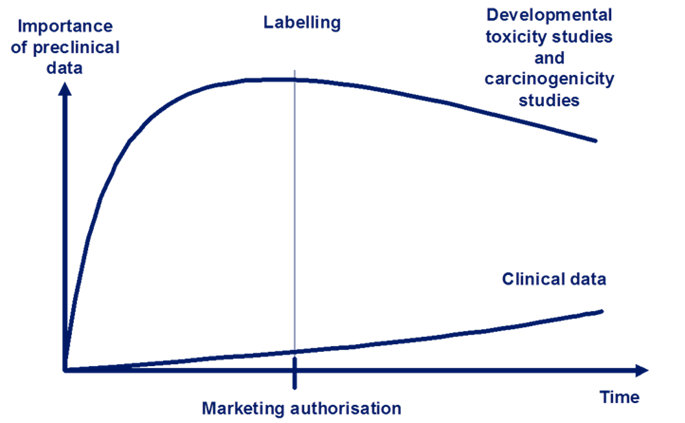
Figure 2: The relative importance and reliance on non-clinical data for human safety. Data from developmental toxicity studies and carcinogenicity studies continue to be relied upon more than clinical data throughout development and after marketing authorisation. Adapted from Non-clinical Assessment Requirements, Maria Nieto-Gutierrez; Safety and Efficacy of Medicines/Human Medicines Development and Evaluation.
3. Non-clinical studies: Ability to predict studies in humans
Historically, there have been challenges as to how suited non-clinical studies are to predict human studies (also referred to as their predictive value). These challenges have been related to pharmacokinetics, pharmacodynamics (efficacy), and safety in humans which are difficult to predict in non-clinical studies. There is a rapid development in this area. Many new technologies in silico (computer models), pharmacogenomics, biomarkers, and new exploratory designs for clinical trials are being developed. They all have a positive influence on the predictive value of non-clinical studies.3.1. Animal Models
The use of relevant non-clinical models and animal species is key to obtain predictive data for humans. This accounts for all types of medicines and clinical trials in the medicine development process. For most new medicines, science-driven strategies are applied. This is especially true when researchers study biologics . Therefore much effort goes into selecting the test systems and animal species that are most suited to predict studies in humans.
Animal species are selected based on how similar the animal species and humans are. It depends on aspects such as:- Pharmacodynamics (safety pharmacology)(what the medicine does to the body).
- Pharmacokinetics (what the body does to the medicine).
- Physiology and pathophysiology.
The pharmacodynamics in the animal species should be similar to that of humans. The target, structural homology (shared ancestry), distribution, communication pathways in the cells, and the effects of the medicine should all be taken into account. Non-clinical trials should collect information on the pharmacokinetics of the drug candidate. This information will be used to calculate the first doses in early clinical trials and to predict the therapeutic doses in later trials. The calculations must be based on the result from toxicology studies and in the case of biologics, the calculations are often based on the body's response to the medicine. When researchers select an animal model, it is important that they evaluate the physiology and pathophysiology of the animal species in question against that of humans. Healthy animals have been used to predict efficacy and safety in patients. However since the patients have a disease, they also have an altered physiology. Therefore, animal models with the disease in question are now often used in non-clinical testing. Special considerations must be taken to deduct or extrapolate data from animals to special groups such as paediatric and geriatric patients, or pregnant women.
The choice of animal species also depends on practical considerations, such as how available they are and how easy they can be used in standard laboratory environments and procedures. Researchers will often do a screening test before the animal species is selected.
Some examples of animal models include:- Rat (osteoporosis, inflammatory diseases, diabetes, obesity, cardiovascular dysfunctions, neurodegenerative diseases, cancers).
- Monkey (osteoporosis, inflammatory diseases).
- Pig (cardiovascular dysfunctions such as hypertension).
- Mouse (cancers, some genetic diseases).
3.2. Specific Animal Models
- Rats and dogs are a common choice of animal model in general toxicity studies (repeated-dose toxicology studies). Except if they are not suited due to pharmacodynamic, pharmacokinetic, and/or pathophysiological differences.
- Rats are often selected for reproductive toxicology studies (that assess the effects on fertility, embryo-foetal development, and pre- and post-natal toxicity).
- Rabbits are often chosen as a second, non-rodent species for studies that assess the potential for embryo-foetal toxicity. Rabbits may be replaced by non-human primates if the rabbits are not suited for the studies which is often the case for biotechnology
products.
- Rats, mice, or hamsters are common in long-term carcinogenicity studies. Transgenic mice can be used in short-term study designs if additional assessment is needed on carcinogenicity.
- Other non-clinical study types address specific aspects of safety, e.g. if people can become addicted to the drug (rodents, primates), vaccines (ferrets), immunotoxicity (mice), hypersensitivity (guinea pig), and dermal, topical toxicity (pig).
- For some studies, the most common animal models can not be used. In these cases, rats can be replaced with hamsters, gerbils, or guinea-pigs and dogs might be replaced by mini-/micro-pigs or monkeys.
Sometimes it is not possible to find a ‘relevant’ and predictive animal species. This is often the case with biologics. In those cases other approaches are recommended. These include the use of relevant transgenic animals which express the human target, or the use of proteins that have the same structural features and gene-patterns (homologous proteins). Researchers are always adviced to include intensive monitoring in protocols. Also, significant safety measurements are needed in the clinical trial setting.
4. Timing of non-clinical studies to the clinical trial programme
- Pharmacology studies.
- General toxicity studies.
- Toxicokinetic and non-clinical pharmacokinetic studies.
- Repeated-dose toxicity studies.
- Assessments of carcinogenic potential.
- Phototoxicity, immunotoxicity, juvenile animal toxicity, etc.
- Biotechnology-derived products (guideline issued under ICH topic S62).
- Life-threatening or serious diseases – such as resistant HIV infection or congenital enzyme deficiency diseases that don’t have a current effective therapy.
- Medicines that use innovative therapeutic modalities (for example, siRNA or vaccine adjuvants) and where non-clinical studies can be shortened, postponed, left out, or added in the non-clinical programme.
The non-clinical
safety assessment programme has more goals. These include:
- To characterise toxic effect.
- To identify target organs.
- To clarify dose dependence.
- To understand how toxicities are related to exposure.
- To assess potential reversibility.
The table below shows the standard non-clinical programme that must be completed before the clinical programme may begin.
|
Type of studies |
Aim |
|
Safety pharmacology core studies |
Assess effects on cardiovascular, respiratory, and central nervous systems (CNS). |
|
Primary pharmacodynamics |
In vivo and/or in vitro studies. Assess mode of action/ |
|
Data gathered during in vitro studies on metabolic and blood protein binding data for animals and humans. Systemic exposure data from toxicology studies (Area under the curve, (AUC)). |
|
|
Acute toxicity studies |
Single-dose toxicity studies in two mammalian species – but can be completed during studies that define a maximum tolerated dose in the species used for toxicity testing. |
|
Vary in length according to duration, therapeutic indication and scope of the proposed clinical programme. Minimum duration for two weeks in two species (one of which is not a rodent). |
|
|
Other studies of concern |
For instance, examination of phototoxicity (when the skin reacts when exposed to light) |
Table 1: Standard non-clinical study programme before ‘first-in-human’ clinical trials. In this phase, single dose, data on lethality or reproductive studies are not needed. Table adapted from ICH (2009) M3(R2).
5. Toxicology Studies
Toxicology studies examine the safety profile of the development candidate. They also provide important information about the absorption, distribution, metabolism, and excretion (ADME) of the active substance in the body. A medicinal product must be assessed in many different kinds of non-clinical toxicology studies before it can beadministered to the first human volunteer. And before the medicine receives a marketing authorisation more toxicology studies are required.
The following gives an overview of the different types of toxicology studies that may be needed in a non-clinical programme. Figure 3: Types of toxicology studies in a non-clinical testing programme.
Figure 3: Types of toxicology studies in a non-clinical testing programme.
5.1. Repeated-Dose Toxicity
The repeated-dose toxicity studies in animals are designed to include a similar or longer exposure time than in human studies. As shown in Table 2, repeated-dose toxicity studies in two species (one non-rodent) for at least two weeks would generally support any clinical trial of up to two weeks. Clinical trials of more than two weeks should be supported by repeated-dose toxicity studies of at least the same duration. Six-months rodent and nine-months non-rodent studies generally support treatment for more than six months in clinical trials.|
Maximum Duration of Clinical Trial |
Recommended Minimum Duration of Repeated-Dose Toxicity Studies to Support Clinical Trials |
|
|
Rodents |
Non-rodents |
|
|
Up to 2 weeks |
2 weeksa |
2 weeksa |
|
Between 2 weeks and 6 months |
Same as clinical trialb |
Same as clinical trialb |
|
Longer than 6 months |
6 monthsb,c |
9 monthsb,c,d |
|
a In the United States, extended single-dose toxicity studies can replace two week studies to support single-dose human trials. b In some circumstances, clinical trials longer than three months can start, if data are available from a 3-months rodent and a 3-months non-rodent study. On a case-by-case basis, this extension can be supported by chronic, in-life and necropsy data. c There can be cases where children are the primary population, and where existing animal studies have identified developmental concerns. In these cases, it can be appropriate to do long-term toxicity testing in juvenile animals. d In the EU, studies in non-rodents for six months are considered acceptable. However, where studies with a longer duration have been conducted, it is not appropriate to conduct an additional study of six months. |
||
Table 2: Recommended duration of repeated-dose toxicity studies to support the conduct of clinical trials. Table adapted from ICH (2009) M3(R2).
The recommended durations of repeated-dose toxicity studies needed to support a marketing authorisation application (MAA) are shown in Table 3 below.
|
Duration of Indicated Treatment |
Rodent |
Non-Rodent |
|
Up to 2 weeks |
1 month |
1 month |
|
More than 2 weeks to 1 month |
3 months |
3 months |
|
More than 1 month to 3 months |
6 months |
6 months |
|
More than 3 months |
6 monthsa |
9 monthsa,b |
|
a There can be cases where children are the primary population, and where existing animal studies have identified developmental concerns. In these cases, it can be appropriate to do long-term toxicity testing in juvenile animals b In the EU, studies in non-rodents for six months are considered acceptable. However, where studies with a longer duration have been conducted, it is not appropriate to conduct an additional study of six months. |
||
Table 3: Recommended duration of repeated-dose toxicity studies to support an MAA. Table adapted from ICH (2009) M3(R2).
Testing in both rodent and non-rodent animals is important to obtain as much data as possible on toxicity for a given medicine. Each "animal type" has features that can be transposed into human situations and that can help researchers understand the potential toxic effect of the medicine in humans.
5.3. Local Tolerance Studies
Local tolerance studies examine how the drug affects the skin or the eyes. These studies are usually part of the general toxicity studies. A single-dose local tolerance study in one species is usually enough to support limited human administration by non-therapeutic routes. An example could be a single intravenous dose for determination of absolute bioavailability.5.4. Genotoxicity Studies
Genotoxicity studies test the effect of the medicine on the chromosomes and genes. They are generally needed to support human safety. Assessment ofgene mutations, is considered enough to support all single-dose clinical trials. For multiple-dose clinical trials, an additionalassessment of chromosomal damage in mammalian systems must be performed. In addition, a full battery of tests for genotoxicity should be completed before Phase II clinical trials. If the test results show genotoxicity, additional testing must be considered.5.5. Carcinogenicity Studies
5.6. Reproduction Toxicity Studies
Reproduction toxicity studies test how the medicine may influence the ability to reproduce and develop normally. How these studies are carried out depends on the study population, and the following considerations:
- Men can be included in Phase I and II clinical trials before the male fertility study, becausethe reproductive organs are evaluated in the repeated-dose toxicity studies. A male fertility study should be completed before initiatinglarge scale or long clinical trials (for instance, Phase III trials).
- Women who are not of childbearing potential (for instance permanently sterilised or postmenopausal women) can take part in clinical trials without reproduction toxicity studies. In this case, the relevant repeated-dose toxicity studies must be completed
beforehand (these studies include an evaluation of the female reproductive organs).
- If women of childbearing potential (or WOCBP) are potential users of the medicine, reproduction toxicity studies need to be done as early as possible.
6. Inclusion of Women of Childbearing Potential in Clinical Trials and Embryo-Foetal Development Toxicity Studies
There is a big concern for unintentionally exposing an embryo or foetus in women of childbearing potential. Such women should not be exposed without intent before data have become available that allow assessing potential benefits and risks. The women can be included in early clinical trials in some situations without non-clinical embryo-foetal developmental toxicity studies. Women do wish to become more involved and in order to do so safely, ways to minimise risks include:
- Reproduction toxicity studies can characterise the risk of a medicine.
- Risks can be limited if pregnancy is prevented during clinical trials. This can be done with pregnancy tests (e.g. based on human chorionic gonadotropin (hCG)) use of highly effective methods of birth control (failure <1%), orstudy entry only after a confirmed menstrual period.
In some cases, it might be difficult to conduct embryo-foetal development toxicity studies in an appropriate animal model (as in some medicines derived from biotechnology). Researchers therefore have more options to consider that can minimise the risks for teratogenicity (causing foetal malformations):
- Inform the participant of possible risks to the embryo or foetus.
- Educate the participant to ensure compliance.
- Test for pregnancy during the trial.
- Know the mechanism of action of the drug and how exposed the foetus will be. For example, for monoclonal antibodies embryo-foetal exposure is low in humans during organogenesis. Based on this, developmental toxicity studies can be done.
It is normal that preliminary data on reproduction toxicity are available from two animal species when women of childbearing potential (WOCBP) are included in clinical trials. In addition, when measures are taken to prevent pregnancy in clinical trials, up to 150 WOCBP may be included and receive the investigational treatment for a relatively short duration (up to 3 months) and before definitive reproduction toxicity testing. This procedure is acceptable because of the very low rate of pregnancy in controlled clinical trials of this size and duration. Also because preliminary studies that are carefully designed can detect most developmental toxicity that could slow or stop enrolment.
Despite the ICH harmonised guideline, different regions have different requirements for embryo-foetal development toxicity studies before WOCBPs can be included into clinical trials.
If pregnant women and children are included in clinical trials, it requires that all the relevant non-clinical data are available. It is also preferred that data on non-pregnant women have been assessed.
7. Setting the 'First-in-Human' Dose
For many innovative medicines, it is acceptable to estimate the safe start dose for humans. However, for some new medicinal products, an estimate may not be enough to predict serious adverse reactions. Researchers should assess the risk factors and mitigation measures and discuss these before human trials begin. The risk factors should also be taken into account on a medicine by medicine basis.
Risk factors may have to do with the mode of action of the medicine. It is therefore relevant to assess previous human exposure with substances that are related to the medicine, the structure of the medicine, and the evidence from animal models of potential toxicity. Other considerations include nature of the target, intensity effects, and the dose-response relationship. There are risk factors that may require special attention, e.g. metabolic pathways, or inter-species genetic differences . Below are some important factors to consider when the first-in-human dose is set:
- All relevant non-clinical data, including:
- Pharmacological dose response studies;
- Pharmacological/toxicological profile;
- Pharmacokinetics studies.
- The No Observed Adverse Effect Level (NOAEL) is the most important information. NOAEL is the highest dose level that does not produce a significant increase in adverse effects in comparison to the control group. In more simple terms, the NOAEL is the highest dose where no important adverse effects are seen.
- The clinical start dose depends on various factors:
- Pharmacodynamics.
- Particular aspects of the molecule.
- Design of the clinical trials
- Exploratory clinical trials in humans can begin with different, nonclinical support. Therefore, the estimation of the clinical start (and maximal) dose can differ.
Other considerations when setting first-in-human dose are:
If researchers gain early access to human data, it can improve the insight into human physiology/pharmacology. It can also improve the knowledge of a drug’s characteristics and its relevance for the therapeutic target and eventually the disease. The term ‘exploratory clinical trials’ (Phase 0) was developed to meet this need. There are different approaches to carry out such exploratory trials:
- They are to be conducted before or early in Phase I.
- They involve limited human exposure.
- They have no therapeutic intent.
- They are not intended to examine clinical tolerability.
There are two classical ways to establish the first human dose in Phase I (I/II) clinical trials.
-
It can be established based on the NOAEL (No Observed Adverse Effect Level) from toxicity studies. A safety factor will have to be taken into consideration as well as individual growth allometric scaling.
- For biologics where their risk factors are identified, it can be established using the Minimal-Anticipated-Biological-Effect-Level (MABEL) standard and also applying a safety factor. To estimate the MABEL, all relevant in vitro and in vivo data are considered. The safety factor is set on the basis of risk criteria. These could for example be how new the active substance is, how potent it is biologically, its mode of action, itsdegree ofspecies specificity, and its dose-response relationship.
The following figure gives a summary of the MABEL approach.

There are other situations, such as clinical trials in cancer patients, where, yet again, other methods may be applied to set the dose.
The Committee for Medicinal Products for Human Use (CHMP) issued a guideline in 2007 on strategies to identify and mitigate risks for first human clinical trials with high risk investigational medicinal products. This guide is recommended to give more information if interested.