Basic principles of non-clinical development
5. Non-clinical regulatory guidelines
There are many players involved in the development of medicines. Also, each of these players (organisations or institutions) has their set of ‘rules’. For instance, companies have their Standard Operating Procedures (SOP). And the European Medicines Agency (EMA) has published many guidelines related to non-clinical development (in addition to the guidelines they have published on Good Clinical Practice (GCP)). These guidelines are in line with the ICH (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use.The main guidelines related to non-clinical development are:
- EMA/CPMP/ICH/286/1995: ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. December 2009.
- EMA/CHMP/ICH/731268/1998. Committee for medicinal products for human use (CHMP) ICH guidelines S6 (R1) – preclinical safety evaluation of biotechnology-derived pharmaceuticals. June 2011.
- EMEA/CHMP/SWP/28367/2007, European Medicines Agency (EMEA) (2007b) Guideline on requirements for first-in-man clinical trials for potential high-risk medicinal products.
The guidelines can be found at
EMA’s website. They are either general or more specific (e.g. specific to required toxicology studies) and they address scientific and technical aspects. The guidelines must be strictly followed
for any new marketing authorisation application (MAA). Also, any deviation from the guidelines must be explained. The MAA will be explained in the following.
Before a new medicine candidate can be sold on the market or prescribed to patients, the pharmaceutical company must submit a MAA to the EMA. The MAA is a large dossier (collection of documents) and its purpose is to obtain approval from the EMA and the European Commission to market a new medicine. The MAA contains all the data and study results that have been produced during the development of the drug.
Data in the MAA are presented according to the so-called CTD (Common Technical Document) format. The CTD includes all the data produced on the Quality, Safety and Efficacy for the medicinal product. These three types of data are key for the regulatory authorities to assess the medicinal product. The CTD is defined by the ICH and it has revolutionised the regulatory review process. This is because it has led to harmonised (similar) electronic submissions and good review practices. It has also made it easier for the pharmaceutical industry, because they will not need to reformat the submission content to different regulatory authorities. The ICH brings together the regulatory authorities and pharmaceutical industry of Europe, Japan, and the US to discuss scientific and technical aspects of medicines registration.
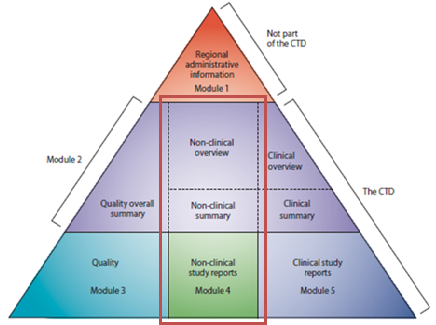
Figure 1: The non-clinical development in CTD modules
Adapted from ICH CTD available at http://www.ich.org/products/ctd.html
The CTD is organised into five parts called modules (Figure 1). Module 1 is region specific and Modules 2, 3, 4 and 5 are setup to be common for all regions. In July 2003, the CTD became the mandatory format for the submission in the EU and in Japan.
From May 2017, it also became mandatory in the US.
The non-clinical development in CTD modules:
- Non-clinical development data/results must be presented in Module 4 of the MAA.
- Non-clinical development summary and overview must be part of Module 2.