Basic principles of non-clinical development
| Site: | EUPATI Open Classroom |
| Course: | Requirements for Non-clinical Studies and the Purpose and Relevance of Animal Testing |
| Book: | Basic principles of non-clinical development |
| Printed by: | Guest user |
| Date: | Thursday, 9 July 2026, 12:05 AM |
Section Overview
- 1. In silico, in vitro, and in vivo studies
- 2. ‘Non-clinical’ or ‘pre-clinical’ studies?
- 3. Key aspects of Chemistry, Manufacturing, Control (CMC) during non-clinical development
- 4. Overview of non-clinical development
- 5. Non-clinical regulatory guidelines
- 6. Non-clinical study types
- 7. First dose estimation in human
- 8. Non-clinical results that can stop the development
- 9. Specifics of non-clinical development for biological medicines
- 10. Conclusions
1. In silico, in vitro, and in vivo studies
(This section is organised in the form of a book, please follow the blue arrows to navigate through the book or by following the navigation panel on the right side of the page.)
There are three different ways to do non-clinical studies:
- ‘In silico’ which means that the study is done on a computer or via computer simulation, e.g. to predict how toxic the medicinal product is based on its chemical structure.
- ‘In vitro’ (Latin for ‘within the glass’) which means that the study is done outside of a living organism and under control, e.g. by use of hepatocyte (cells from the liver) cultures for metabolism studies.
- 'In vivo’ (Latin for ‘within the living’) which means that the study is carried out using a whole, living organism in stead of tissues or cells, i.e. animals, humans or plants.
2. ‘Non-clinical’ or ‘pre-clinical’ studies?
The terms ‘non-clinical’ and ‘pre-clinical’ are often both used to describe the same type of studies. However, EUPATI has decided to only use the term ‘non-clinical’ in all its course materials because this term is seen as more inclusive.
Non-clinical studies can be carried out at any time during a product’s life (product life cycle). However, they should be done as early as possible in order to avoid surprises later on in the development of the new medicine.
Non-clinical studies can be designed to:
- Identify a medicine’s ‘pharmacodynamics’ (what a medicine does to the body).
- Identify a medicine’s ‘pharmacokinetics’ (what the body does to the medicine).
- Determine a medicine’s ‘toxicology’ (how poisonous or toxic the drug is before giving it to humans).
- Determine a medicine’s safety. This can be done by refining, combining and adding information on safety. And it can be done in the non-clinical phase; at the time of product registration or during the product’s life cycle.
3. Key aspects of Chemistry, Manufacturing, Control (CMC) during non-clinical development
Adequate active drug substance must be produced for all non-clinical studies:
- Small quantities (milligrams to grams) are usually needed for non-clinical studies. Later on in clinical studies and for the market, the production will then be scaled up to produce larger quantities.
- For Good Laboratory Practice (GLP) studies, the drug substance must be qualified or follow Good Manufacturing Practice (GMP) for each batch produced.
Some key CMC steps during the non-clinical phase are:
- To determine the dose and administration (how the medicine is given, e.g. the dose or how often it is given).
- To describe the physico-chemical characteristics (traits) of the medicinal product in detail.
- To test the stability & purity of the medicinal product.
- To develop and validate the methods used to quantify the active substance in body fluids such as blood, plasma, or urine. This is done in activity and side effect studies.
- To develop a prototype of the medicine which will be used in the clinic.
4. Overview of non-clinical development
Non-clinical development is carried out in parallel with research activities. It should support the planned clinical development programme and address the questions below.4.1. Objectives of non-clinical development
Non-clinical development should start after a lead development candidate is found. It should answer the following questions, and the answers will come from specific assessments/studies:
- Does the medicine “work”? → the answer will come from efficacy assessments or efficacy studies.
- How can the medicinal product be delivered? And how does the body react to it? → these answers will come from the ADME studies ( Absorption, Distribution, Metabolism and Excretion).
- Is the medicine safe? → toxicology/safety pharmacology studies will provide the answer.
- Is the production (manufacture) of the medicinal product viable and can it be controlled?
As mentioned, non-clinical development activities can continue throughout the life cycle of the product.
4.2. Project management and the Target Product Profile (TPP)
The non-clinical development programme is complex so it requires solid project management and communication skills. Project teams must be setup with specialists that have different expertise and they must understand the clinical plan in order to define the non-clinical plan and related activities.
The Target Product Profile (TPP) is a key document that describes the non-clinical development strategy. It defines the goals, risks, liabilities, metrics and Go/No-Go decision milestones.
The TPP usually contains:
- Expected therapeutic indications and clinical use, key clinical trial endpoint.
- Patient age range, dose route and form, frequency of administration.
- Medicines target, mechanism of action, bioavailability, duration of action.
- Safety, precautions and contraindications.
- Chemistry, manufacturing and controls (CMC).
- Market evaluation.
- Patent status and conditions of exclusivity (e.g. orphan drug status).
The TPP will be updated and should be reviewed from time to time during the medicines development. For example, the project team should evaluate any results that may lead to concerns on safety or the ability to fulfil the criteria in the TPP. The team will also evaluate how the results may impact the success of the project. The TPP helps the project team to focus on key product criteria and to make 'Go/No-Go' decisions in due time. It will also help to reduce the overall project risk.
5. Non-clinical regulatory guidelines
There are many players involved in the development of medicines. Also, each of these players (organisations or institutions) has their set of ‘rules’. For instance, companies have their Standard Operating Procedures (SOP). And the European Medicines Agency (EMA) has published many guidelines related to non-clinical development (in addition to the guidelines they have published on Good Clinical Practice (GCP)). These guidelines are in line with the ICH (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use.The main guidelines related to non-clinical development are:
- EMA/CPMP/ICH/286/1995: ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. December 2009.
- EMA/CHMP/ICH/731268/1998. Committee for medicinal products for human use (CHMP) ICH guidelines S6 (R1) – preclinical safety evaluation of biotechnology-derived pharmaceuticals. June 2011.
- EMEA/CHMP/SWP/28367/2007, European Medicines Agency (EMEA) (2007b) Guideline on requirements for first-in-man clinical trials for potential high-risk medicinal products.
The guidelines can be found at
EMA’s website. They are either general or more specific (e.g. specific to required toxicology studies) and they address scientific and technical aspects. The guidelines must be strictly followed
for any new marketing authorisation application (MAA). Also, any deviation from the guidelines must be explained. The MAA will be explained in the following.
Before a new medicine candidate can be sold on the market or prescribed to patients, the pharmaceutical company must submit a MAA to the EMA. The MAA is a large dossier (collection of documents) and its purpose is to obtain approval from the EMA and the European Commission to market a new medicine. The MAA contains all the data and study results that have been produced during the development of the drug.
Data in the MAA are presented according to the so-called CTD (Common Technical Document) format. The CTD includes all the data produced on the Quality, Safety and Efficacy for the medicinal product. These three types of data are key for the regulatory authorities to assess the medicinal product. The CTD is defined by the ICH and it has revolutionised the regulatory review process. This is because it has led to harmonised (similar) electronic submissions and good review practices. It has also made it easier for the pharmaceutical industry, because they will not need to reformat the submission content to different regulatory authorities. The ICH brings together the regulatory authorities and pharmaceutical industry of Europe, Japan, and the US to discuss scientific and technical aspects of medicines registration.
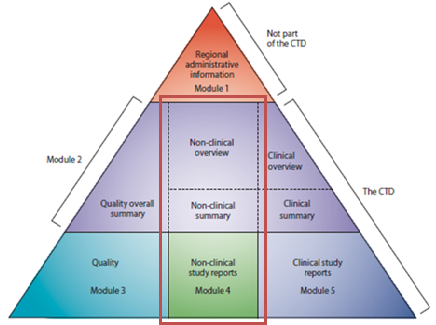
Figure 1: The non-clinical development in CTD modules
Adapted from ICH CTD available at http://www.ich.org/products/ctd.html
The CTD is organised into five parts called modules (Figure 1). Module 1 is region specific and Modules 2, 3, 4 and 5 are setup to be common for all regions. In July 2003, the CTD became the mandatory format for the submission in the EU and in Japan.
From May 2017, it also became mandatory in the US.
The non-clinical development in CTD modules:
- Non-clinical development data/results must be presented in Module 4 of the MAA.
- Non-clinical development summary and overview must be part of Module 2.
6. Non-clinical study types
There are three types or groups of non-clinical development studies:
- Pharmacodynamics (PD) studies.
- Pharmacokinetics (PK) studies.
- Toxicology studies.
6.1. Pharmacodynamics (PD)
Pharmacodynamics studies aim to address - these studies are explained below:
- Primary pharmacodynamics.
- Secondary pharmacodynamics.
- Safety pharmacology.
- Pharmacodynamic interactions.
Primary pharmacodynamics
The goal of primary PD studies is to find the mode of action or effects that are related to the desired therapeutic target (efficacy). These studies:
- Use one or more pharmacological animal models of the disease.
- Help find the best candidate for further development.
- Help select doses for both non-clinical and clinical studies.
The studies can be done in-vivo and/or in-vitro. They are generally conducted during the discovery phase and are usually non-GLP (not done according to Good Laboratory Practice (GLP)).
Secondary pharmacodynamics
The goal of secondary PD studies is to find the mode of action or effects that are not related to the desired therapeutic target. Secondary PD studies may not be needed if literature can give the necessary information instead.
Safety pharmacology
- The objective is to identify unwanted effects on key physiological functions within the therapeutic dose range and higher. Usual studies look into respiratory, central nervous system (CNS), and cardiovascular functions. Studies on respiratory and CNS (e.g. functional observational battery (FOB)) functions are usually performed on rats (in vivo).
- Studies on cardiovascular function are performed in vitro (e.g. human ether-a-go-go related gene (hERG)) and in-vivo (e.g. dogs with telemetry).
- Follow-up studies may be needed if concerns arise.
These studies must be conducted following Good Laboratory Practice (GLP). To reduce the use of animals, the testing should be done in vitro where this is possible.
6.2. Pharmacokinetics (PK)
Pharmacokinetic studies aim to address:- ADME: A (absorption), D (distribution), M (metabolism), E (excretion).
- Toxicokinetics (how much of the medicine is in the body and where/when unwanted effects happen).
6.3. Toxicology
The toxicology studies aim to address how toxic the medicinal product is:- Single-dose toxicity.
- Repeated-dose toxicity.
- Genotoxicity (damage within a cell that causes changes or mutations in the genes).
- Carcinogenicity (can it cause cancer?).
- Development and reproductive toxicity (DART)
- When researchers have a special cause for concern or when the medicine is for long-term use, then the researchers must assess the risk of developing cancer (the drug’s carcinogenic potential).
- Other non-clinical studies should be carried out on a case-by-case basis, as appropriate. These include for example phototoxicity studies, immunotoxicity studies, juvenile animal toxicity studies and abuse potential studies.
Single dose and dose-range finding (DRF) studies
First, these studies are done in rodents (mice or rats), and then in a larger animal species (for example dogs and usually non-primates).
The purpose of the studies is to find out how toxic the developmentcandidate is. This is based on:- The maximum tolerated dose (MTD). The MDT is the highest dose that does not cause unacceptable side effects.
- The non-observed adverse effect level (NOAEL). The NOAEL is the highest dose level that does not produce a significant increase in adverse effects in comparison to the control group.
- The target organ(s) in which toxicity is seen.
- The doses for future toxicology studies or first-in-human studies.
In the DRF/toleration studies, the animals are monitored as the dose is increased and until the dose is found where adverse effects are first seen in one or few animals. Such studies will help define the so-called toxicological dose–response profile (usually for the first time for a medicinal product in development). The studies will include cage side observations (for physical and behavioural effects), drug exposure analysis, blood chemistry, haematology (the study of blood and blood-forming tissues), pathology (the study of the causes and effects of disease or injury mostly through analysis of tissue, cell, and body fluid samples) and histopathology (the study of diseased cells and tissues using a microscope).
Repeated dose toxicology studies
A repeated dose toxicology study will often be carried out in specific animal species. The purpose of this is to confirm the dose levels that will later be used in GLP toxicology and Safety Pharmacology studies.
In general, repeated dose toxicity studies shall be done in two species of mammals. One of these species must be a non-rodent.
The goal is to:- Find the toxicity profile (MTD, NOAEL) when given repeatedly over a period of time.
- Identify target organ(s) in which toxicity is seen.
- Find out if adverse effects can be reversed.
- Find the dose(s) for future toxicology studies or clinical trials.
Their standard duration is:
- Sub-chronic: 7, 14 and 28 days and 3 months
- Chronic: 6, 9 and 12 months.
Genotoxicity studies
The purpose of these studies is to find out if the medicinal product potentially affects DNA or chromosomes because this may lead to gene mutation and/or chromosomal damage.
The standard genotoxicity tests (or test battery) include:- In-vitro bacterial mutation (Ames test).
- In-vitro mammalian cell chromosomal aberration (e.g. Human peripheral blood lymphocytes HPBL).
- In-vivo mammalian cell chromosomal aberration (e.g. rodent micronucleus).
Carcinogenicity studies
Carcinogenicity studies include:- 2-year mouse or 26-week transgenic mouse (a mouse where the genes have been changed by inserting genes from another species).
- 2-year rat bioassay.
Development and reproductive toxicology studies
Development and reproductive toxicology (DART) studies include:- Fertility (typically rat)
- Teratology (typically rat and rabbit)
- Peri- and post-natal (typically rat)
7. First dose estimation in human
8. Non-clinical results that can stop the development
- A development candidate is found toxic in a target organ in animals, e.g. if it is hepatotoxic (toxic for the liver).
- Further development may be questioned. The development may be stopped or possible toxicities will be monitored in clinical trials (although the predictive value of animal studies may still be questioned).
- Poor pharmacokinetic (PK) properties have been found. An example is if a product does not get to its target (poor bioavailability). Other examples are if the medicine builds up, or causes toxicity, or if so-called toxic metabolites (substances that are the result of metabolic processes in the body) are produced.
This also explains why early ADME studies are performed. They can help researchers optimise selection of successful product candidates.
Animal studies that are not designed or reported well may produce bias in the research record. Also, there is a risk that researchers may initiate clinical trials when this is not appropriate. Or they may fail to protect humans from toxic drugs.
9. Specifics of non-clinical development for biological medicines
Biological medicines (or simply biologics) are complex in comparison with small molecules. Biologics are for example large molecules, tissues, cells, or proteins whereas most small molecule medicines are chemically synthesised. The test principles for both drug types are the same but the non-clinical development plan for biologics follows a case-by-case approach.
It should take into account that:- Biologics are often highly species specific, e.g. their pharmacological activity may only be shown in non-human primates (NHP). Also, standard species for toxicity testing may not be relevant.
- Biologics have a strong immunological response. Therefore, there is often no scientific reason to do long-term studies over several months.
- Biologics are usually immunogenic. Immunogenicity assessment can therefore help researchers interpret the animal toxicology data (not done to predict immunogenicity in humans).
- The probable dose level leading to a minimal anticipated biological effect (MABEL) in humans needs to be determined.
Standard development plans start to appear for biologics and are different from those well established for small molecules. These plans are driven by lessons learned from past experience and new regulatory guidance.
10. Conclusions
The non-clinical development phase is critical and must foresee any possible problems before a medicine candidate moves to the clinical development phase (clinical studies done in people).
Before a medicine can be tested in clinical studies the following must be ensured:- Non-clinical safety is evaluated under GLP condition.
- Manufacturing of both, active substance and medicinal product, is executed under proper quality control.
- Data and process are documented according to CTD format. And they build the foundation for the clinical development phase.
Researchers tend more and more to design medicine-like properties in-silico, and also to use bioinformatics methods for modelling and prediction.
The non-clinical process is complex for different reasons:- The use of new pathology models, which include transgenic animals, can aid some stages of preclinical studies. There is a a lack of biomarkers for organ toxicity which is a serious drawback of the present system. A biomarker is a molecule found in body fluids, for example in blood, or in tissues. It is a sign of an abnormal or normal process, a disease or a condition. It can be used to see how well the body responds to a treatment. The lack of biomarkers is especially evident within myocardial infarction tissue damage, liver toxicity and nephrotoxicity.
- The project development teams must coordinate in an effective and timely manner.
- The project teams usually develop a TPP to frame the non-clinical development programme and thereby to focus on key medicinal product criteria as well as to take Go/NoGo decisions in a timely manner.
- Project management requires scientific, technical, legal and regulatory knowledge.
The main regulatory authorities (The EMA in Europe; the FDA in the US and the PMDA in Japan) keep harmonising or unifying the guidelines for non-clinical development, and they focus on safety and quality. The ICH (www.ich.org) issues detailed guidance from time to time for the pharmaceutical industry. These are similar to the guidelines that are published by the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA).